Ovary Because of the high mortality rate associated with ovarian carcinoma it
is evident that research into novel therapeutic strategies, such as gene
therapy, will be required if meaningful reductions in death caused
by this disease are to be realized. Localization of ovarian cancer cells
within the peritoneal cavity favors concentration of gene therapy
vectors within this container to achieve an optimal therapeutic effect. Moreover, ovarian tumor cell
lines and primary ovarian cell cultures have been shown to be transducible
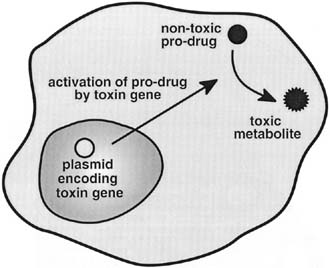
by gene therapy vectors in current use. The toxin commonly used for ovarian carcinoma to accomplish molecular chemotherapy
is the TK gene from the HSV. Experimental evidence has documented
the ability of HSV-TK/GCV-based gene therapy
to mediate tumor cell death and significantly improve survival in orthotopic
murine models of intraperitoneal ovarian cancer.38 Mutation compensation or gene replacement strategies have also been extensively
investigated in the context of ovarian carcinoma. Early examples
of tumor suppressor gene replacement in ovarian carcinoma focused
on the p53 gene. Janicek and associates, using antisense oligonucleotides targeted
to p53 and c-myc, demonstrated growth inhibitory effects that functioned synergistically
in some ovarian carcinoma cell lines.39 Wolf and associates then demonstrated that ovarian cancer cells are growth-inhibited
by transfection with adenovirus-mediated p53 regardless of their endogenous p53 status.40 Additional utility of gene therapy may be found in its ability to enhance
treatment of ovarian cancer cells with conventional chemotherapeutic
agents. To this end, adenoviral mediated delivery of p53 has been demonstrated to sensitize ovarian cancer cells to paclitaxel and cisplatin in both in vitro and in vivo models of ovarian carcinoma.41,42PTEN and p16 are cell cycle regulatory genes that have been delivered by adenoviral-mediated
gene transfer and resulted in growth inhibition in ovarian
cancer cell lines.43,44Bax represents a member of the pro-apoptotic Bcl gene family. Delivery of adenovirus containing Bax gene constructs have been shown to induce ovarian tumor cell cytotoxicity
and apoptosis in in vitro cell culture, as well as nude mouse models of ovarian carcinoma.45 Further utility of Bax-mediated gene therapy also may lie in its ability to sensitize
ovarian cancer cells to the effects of ionizing radiation.46 A widely explored cell surface tyrosine kinase target is the erbB-2 protein
product of the HER2-neu gene. This gene is frequently amplified in many human cancers, including
ovarian carcinoma. Xing and associates have demonstrated prolonged
survival in a mouse model of ovarian carcinoma using liposome-mediated
gene transfer of the K1 gene, which produces the SV40 large T antigen known to suppress cell surface
erbB-2 expression.47 Alternatively, Ueno and associates. have targeted down-regulation
of the erbB-2 protein by liposome-mediated delivery of
the E1A gene.48 These authors have noted significant ovarian tumor cell growth inhibition in vitro and prolonged survival in animal models of ovarian carcinoma. Moreover, these
authors have demonstrated that E1, a mediated down-regulation
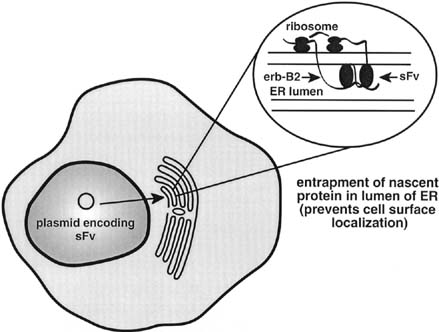
of erbB-2, can sensitize cells to the toxic effects of paclitaxel chemotherapy.48 Single-chain antibodies delivered by adenoviral vectors and directed
against cell surface erbB-2 have also resulted in tumor cell
cytotoxicity and prolonged survival in animal models of ovarian cancer.49 Additionally, adenoviral-mediated gene transfer of anti-erbB-2 single-chain antibodies have been shown to sensitize
erbB-2 overexpressing SKOV-3 ovarian cancer cells to
the toxic effects of cisplatin.50 Gene-based strategies to enhance the immune response in ovarian
carcinoma has focused on increasing local concentrations of cytokines, as
well as up-regulating the presence of co-stimulatory
molecules on the surface of ovarian cancer cells. In vitro studies of AAV-mediated IL-2 gene transfers have resulted
in augmented local concentrations of IL-2 in ovarian cancer cell
line cultures.51 Xu and associates have demonstrated prolonged survival with increased
nitric oxide production after implantation of an ovarian cancer cell line
transfected with the interferon beta gene into the peritoneal cavity
of nude mice.52 Additionally, Son used liposome-mediated gene transfer of the interferon
gamma gene in a murine model of metastatic ovarian carcinoma
to prolong survival and inhibit tumor nodule growth.53 Finally, Gilligan and associates demonstrated enhanced immunogenicity
of ovarian tumor cells adenovirally transduced to express the co-stimulatory
molecule, B7-1.54 Cervix Two HPV early genes, E6 and E7, play critical roles in the development
and maintenance of a malignant phenotype and progression of disease. Several
gene therapy strategies targeting E6 and E7 have been developed. Howley
and others have shown that inactivation of p53 and Rb with the E6 and E7 oncoproteins, respectively, are key steps in the development
of cervical cancer and immortalization of cervical epithelial
cells.55 In light of these findings, Hamada and colleagues sought to explore the
effects of a wild-type p53 recombinant adenoviral vector on cell growth in cervical carcinoma cells.56 In their study of HeLa cells, cell growth of infected HeLa cells was significantly
suppressed in vitro, and the p53 protein was detected in these cells by both Western blot
and immunohistochemistry. In a subsequent study, they were able to demonstrate
that treatment of subcutaneous nodules in nude mice with this
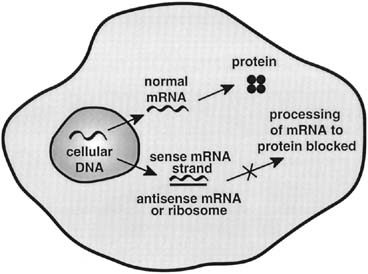
vector resulted in significant volume reduction.57 Von Knebel Doeberitz and others were able to demonstrate that expression
of antisense HPV-18 E6 and E7 RNA could inhibit the growth of
the cervical cancer cells.58 Further exploring the usefulness of directing strategies against HPV E6 and
E7 oncogenes, Hamada and others studied the effects of introducing
an antisense RNA transcript of E6 and E7 genes into cervical cancer
cells.59 After construction of a recombinant adenoviral vector containing a cytomegalovirus
promoter along with antisense RNA oligonucleotides of HPV16 E6 and
E7 genes, they tested its effects on cell growth on the SiHa
cervical cancer cell line, which is positive for HPV16 and contains wild-type
p53. Their results indicated that these antisense transcripts
suppressed growth of SiHa cells in a dramatic fashion. Similarly, in
a study by Cho and associates, they were also able to demonstrate
apoptotic changes in CaSki and SiHa cells after transfection with a
plasmid containing HPV16 antisense nucleic acid.60 Moreover, molecular indications of apoptosis such as loss of mitochondrial
transmembrane potential, release of mitochondrial cytochrome c into
the cytoplasm, and activation of caspases 3 and 9 were seen. P21, which is overexpressed in senescent fibroblasts, has been shown to
bind and inhibit cyclin-dependent kinases, which are required
for cell cycle activity.61,62 It has been recently suggested that p21 also can induce apoptotic cell
death, as seen in vascular smooth muscle cells and retinoblastoma cells. In
a recent study by Tsao and associates, multiple HPV-negative
and positive human cervical cancer cell lines were infected with
a recombinant adenovirus containing p21 cDNA.63 Massive cell death via apoptosis was observed in all cervical cell lines
infected with this novel vector, which was most likely secondary to
p21 transgene overexpression. Bax, a member of the bcl-2 family, potently induces apoptosis via caspase-dependent
and independent mechanisms and is a key downstream
component of the p53 apoptotic pathway.64,65,66 Additionally, it has been shown that bax can function as a tumor suppressor.67 In work performed by Huh and others, multiple cervical cancer lines, including
HeLa, C33A, and CaSki, and primary human cervical cancer cells
lines were infected with a recombinant adenoviral vector containing
the bax gene.68 Potent cytotoxicity via apoptosis and high transduction efficiency was
observed in all three cervical cancer cell lines. Furthermore, primary
human cervical cancer cells were isolated and infected with Ad/Bax
and Ad/Cre; again, robust cytotoxicity via an apoptotic pathway
was noted. Wildner and others tested the efficacy of a replication competent adenovirus (Ad.TKRC) with the HSV-TK gene in nude mice with
subcutaneous nodules of ME180 cervical carcinoma cells.69 Treatment of tumors with Ad.TKRC resulted in an antitumor response similar
to that achieved by a replication-deficient Ad.TK with GCV. This
finding indicated that Ad.TKRC alone without GCV has definitive
oncolytic activity. There was no difference in antitumor effect between
Ad.TK with GCV, Ad.TKRC alone, and Ad.TKRC followed by GCV 24 hours
later. However, when GCV was administered 72 hours after administration
of Ad.TKRC, survival was enhanced, with 60% of mice surviving
more than 160 days. These data emphasize the importance of viral amplification
and the conversion of transduced cells into adenoviral production
cells. In a study by Bilsland and others, they evaluated a novel
adenoviral suicide gene therapy vector expressing bacterial nitroreductase.70 By exploiting telomerase promoters in cervical cancer cells, they were
able to use the bacterial nitroreductase gene that bioactivates the prodrug
CB1954 into an active cytotoxic alkylating agent. Infection with
adenoviral telomerase-NTR constructs resulted in an efficacious
effect in telomerase-positive cervical cancer cell lines and
cervical xenograft models.70,71 Some tumor cells fail to be immunogenic because of the absence of co-stimulatory
molecules. Kaufmann and associates evaluated whether
HPV16-positive cervical cancer cells transfected with the CD80 gene could elicit a HPV16 E7-specific cytotoxic T lymphocyte response
in humans.72 Their work was the first to demonstrate that cervical cancer cells expressing CD80 were able to stimulate cytotoxic T lymphocytes using HPV16 E7 as tumor-associated
antigens. In another work by Gilligan and associates, they
were able to create a recombinant E1/E3-deleted adenovirus
encoding the B7-1 co-stimulatory molecule.73 Cells expressing the B7-1 antigen were noted to have a higher level
of T cell proliferation compared with tumor cells modified by a control
virus containing the β-galactosidase gene. Thus, based
on their evidence, adenoviral-mediated delivery of B7-1 to
tumor cells may have some usefulness in the setting of immunopotentiation
and, ultimately, the development of tumor vaccines by this route. Finally, Dall
and others74 were able to construct a recombinant retroviral vector containing a single-chain
antigen-binding fragment that recognizes specific
epitopes that are frequently detected in cervical cancer but not
in normal cervical epithelium.75 Using the aforementioned vector, they transferred the gene into a murine
cytotoxic T cell line. They demonstrated that all recombinant clones
were able to express the protein on the cell surface, and significant
cytotoxicity was noted in cells expressing these epitopes in a MHC-independent
manner. In a recent study by Liu and associates, dendritic cells were pulsed with
adeno-associated virus with the HPV16 E6 gene.76 This approach revealed transduced E6 gene mRNA expression along with chromosomal
integration in infected dendritic cells. Rapid induction of
a cytotoxic T cell response against primary cervical cancer cell lines
was seen. Uterus At the present time, the usefulness of gene therapy in the setting of endometrial
cancer is primarily based in in vitro studies. In a study by Kunishige and associates, the HSV-tk gene was transfected into the endometrial adenocarcinoma cell line, HHUA, via
a nonviral vector.77 They were able to demonstrate a bystander effect and significant growth
inhibition of HSV-tk-negative cells when the population
of cells contained more than 3% HSV-tk-positive
cells. In another study by Niu and colleagues, they were able to transfer the
thymidine kinase gene via a nonviral vector to cultured human and rat
uterine leiomyoma cells.78 Again, a bystander effect was seen in leiomyoma cells transfected with
the thymidine kinase gene and treated with ganciclovir. Forty-eight
percent to 65% of cell death was witnessed when only 5% of
leiomyoma cells were transfected with the TK gene. It also appeared that estradiol enhanced the bystander effect in rat leiomyoma cells. Similarly, in a
study by Ural and colleagues, retroviral-mediated transfer of the HSV-tk gene was studied in the endometrial cancer cell line, EC4.79 With administration of ganciclovir, cell death and inhibition of proliferation
was seen in vitro, along with a significant bystander effect. Replacement of wild-type PTEN, a frequently altered gene in endometrial carcinomas, was evaluated by
Sakurada and associates.80 Using a recombinant adenoviral vector, replacement of wild-type PTEN in endometrial cancer cell lines with inactivated PTEN resulted in decreased cell growth in vivo via apoptotic induction in addition to complete suppression of ex vivo tumor formation. Gene therapy approaches in uterine papillary serous carcinomas, an
aggressive variant of endometrial carcinomas and histologically
similar to papillary serous carcinomas of the ovary, has been
studied as well. Ramondetta and associates studied the efficacy of adenoviral
vectors containing p53 and p21 in a uterine papillary serous carcinoma
cell line (SPEC-2) that contains a mutated form
of p53.81 Both adenoviral vectors decreased cell proliferation; however, the effects
of the adenoviral vector containing p21 were not as significant and
were more transient when compared with the p53 vector. As such, the
authors suggest that perhaps the p53 vector may be a more effective gene
therapy agent in this subgroup of endometrial carcinomas. |