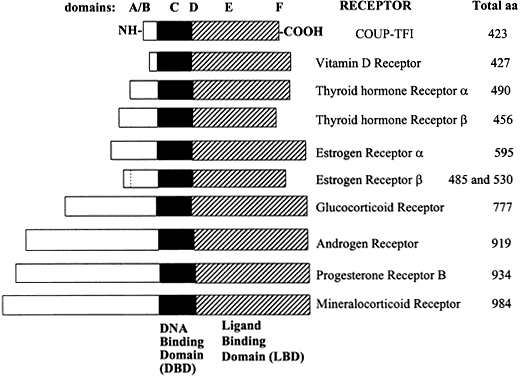
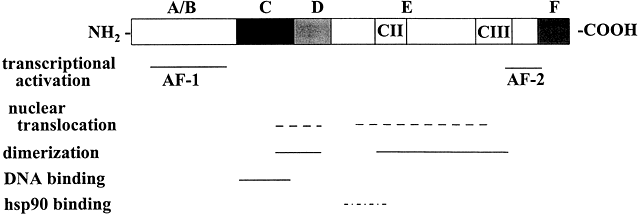
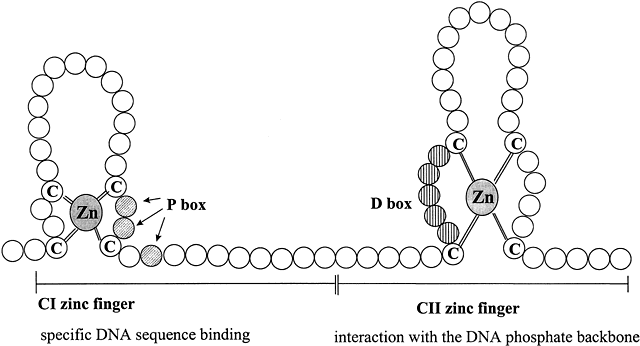
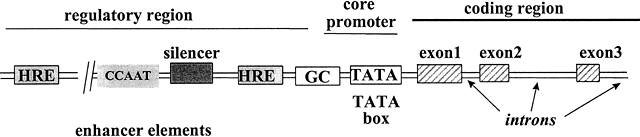
Arrival and Entry of Steroid Hormones Into Target Tissue Cells
Steroid hormones are small, hydrophobic, and lipid-soluble molecules
derived from cholesterol. They circulate in blood free or bound (95%)
to plasma carrier proteins.56,57,58
Sex hormone-binding globulin (SHBG), also known as testosterone-estradiol-binding
globulin (TeBG), and androgen-binding protein (ABP) are encoded by the
same gene.59 ABP is produced by the Sertoli
cells of the testis, and SHBG is produced by the liver and is present
in the circulatory system.58 SHBG binds
most gonadal steroids, and corticosteroid-binding globulin (CBG or transcortin)
binds glucocorticoids and progesterone with different affinities. When
circulating levels of steroid hormones exceed the binding capacity of
their respective binding proteins, they can then bind nonspecifically
and with low affinity to albumin, from which they can readily dissociate
and enter target cells.60 The unbound and
loosely albumin-bound steroids are believed to be the most biologically
important fractions because the steroid is free to diffuse or be actively
transported through the capillary wall and lipid plasma membrane bilayer.
Extracellular binding proteins may modulate hormone response by regulating
the amount of steroid available to the cell.60
The binding capacity of binding globulins is influenced by endocrine status
and other factors.57
SHBG binds to a specific cell membrane receptor called sex hormone-binding
globulin receptor (SHBG-R) and activates adenylyl cyclase, increasing
intracellular cAMP.58 Binding of SHBG to SHBG-R also transfers SHBG into the cell as a consequence
of receptor-mediated endocytosis.61 The interaction of SHBG with SHBG-R is inhibited when steroids are bound
to SHBG, suggesting that SHBG is an allosteric protein.61 However, if unliganded SHBG is allowed to bind to its receptor on intact
cells and an appropriate steroid hormone then is introduced, adenylate
cyclase is activated and intracellular cAMP increases.62 After the steroid hormone is in the cytoplasm, it is not clear whether
a transport protein is required for movement of the hydrophobic steroid
molecule through the aqueous cytoplasm to the receptor, regardless
of whether the receptor is cytoplasmic or nuclear in location. The currently
accepted model is that the steroid hormone diffuses freely in the
cytoplasm. Intracellular Localization of the Steroid/Nuclear Receptors Early attempts to isolate steroid hormone receptors led to a major controversy
regarding their intracellular localization in the unliganded state. Reports
published before 1984 suggested that, before hormone treatment, steroid
receptors in target tissues were located predominantly
in the cytosolic fraction as large protein complexes of 300 to 400 kd. After
hormone exposure, receptors were detected primarily in the nuclear
fraction.63 It was initially proposed that unoccupied and untransformed receptors
were located in the cytoplasm until ligand binding, which caused their
translocation into the nucleus. The reported presence of ERs and PRs
in cytosol is now acknowledged to be largely artifactual, the result of
homogenization and centrifugation processes used to isolate the receptor
protein.64 The newly synthesized unliganded receptor is highly unstable and moves
into the nucleus, targeted by its NLS, or associates with the HSP90 complex
of cytoplasmic proteins.65 Although newly synthesized receptor proteins would be expected to contribute
a small amount to cytoplasmic levels of receptor, most steroid/nuclear
receptors in the unliganded state reside in the nucleus. The exceptions
are the GRs and MRs, which in the unliganded state reside in
the cytoplasm in a complex containing HSP90, HSP70, and a variety of
receptor-associated proteins.66 HSP90 is a general molecular chaperone involved in the folding of various
proteins. The HSP90 complex of proteins has chaperonin activity that
facilitates hormone binding and subsequent proper folding of the GR.67 The HSP90 dimer is thought to stabilize the receptor, protecting it from
protease degradation; block the DBD and the NLS; and maintain the receptor
in an inactive state until ligand binding occurs (Fig. 5).68 HSP90 is also required for ligand binding by the steroid receptors.66 In addition to HSP90, unliganded steroid receptors extracted from animal
tissues or mammalian cells showed that GRs and PRs are complexed with
a number of other proteins, including HSP70, FKB59, p60, p48 (Hip), and
p23.66,67 These proteins are thought to be required for the assembly and maintenance
of ligand-sensitive aporeceptor complexes. 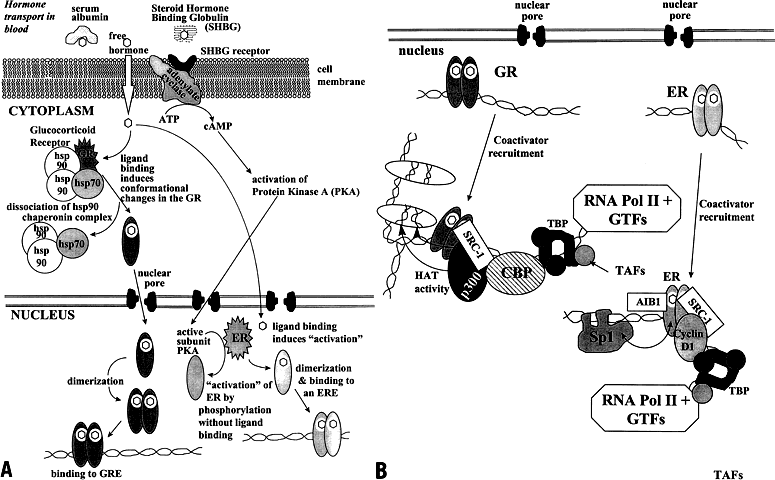 Fig. 5. Mechanism of steroid hormone receptor action in a target cell. A target
cell contains the steroid hormone receptors required to respond to a
given steroid hormone. A. Most steroid hormones that enter the cell travel in the bloodstream in
free state or loosely bound to serum albumins. Estrogens and androgens
are bound to steroid hormone-binding globulin (SHBG) with high affinity. The
cell membrane contains a SHBG receptor linked to adenylate cyclase. Activation
of adenylate cyclase by the SHBG receptor or peptide
hormones such as luteinizing hormone generate cAMP, which activates the
protein kinase A (PKA) phosphorylation cascade. PKA can activate the
estrogen receptor-α (ERα) and other steroid hormone receptors
in the absence of ligand binding. Most receptors in the steroid/nuclear
receptor superfamily reside in the nucleus in the newly synthesized
or unliganded state. The exception is the glucocorticoid receptor (GR), which
is complexed with the HSP90 chaperonin complex in the cytoplasm. HSP90 and
its accompanying proteins are thought to stabilize the
receptor until hormone binding occurs. Although receptors for progesterone, estrogens, and
androgens have also been found in complexes with
the primarily cytosolic HSPs, the function of these associations is unclear. In
the case of estrogens, progestins, and androgens, the hormones
freely enter the nucleus and bind to their cognate receptor. Binding
of hormone ligand to the receptor activates the receptor by inducing
alterations in the conformation of the receptor protein. This allows
the receptors to form homodimers and bind to specific hormone response
elements (HREs) (see Table 2) in the DNA in the regulatory region of the target gene. B. Once bound to the HRE, the liganded steroid hormone receptor induces a
DNA bend and, in the presence of agonist ligand, recruits coactivator
proteins. The HRE-bound receptor also interacts with other DNA-bound
transcription factors, as shown for the interaction of Sp1 with ER. Certain
coactivator proteins have histone acetyltransferase (HAT) activity that acetylates lysine residues in histones H3 and H4. This
results in a "loosening" of chromatin structure that facilitates the
binding and transcriptional activity of RNA polymerase II at the transcription
start site. (Adapted from Naar Am, Beaurang PA, Robinson KM
et al: Chromatin, TAFs, and a novel multiprotein coactivator are required
for synergistic activation by Sp1 and SREBP-1a in vitro. Genes Dev 12:3020–3031, 1998; and from Zwijsen RML, Buckle RS, Hijmans
EM et al: Ligand-independent recruitment of steroid receptor coactivators
to estrogen receptor by cyclin D. Genes Dev 12:3488–3498, 1998.) Fig. 5. Mechanism of steroid hormone receptor action in a target cell. A target
cell contains the steroid hormone receptors required to respond to a
given steroid hormone. A. Most steroid hormones that enter the cell travel in the bloodstream in
free state or loosely bound to serum albumins. Estrogens and androgens
are bound to steroid hormone-binding globulin (SHBG) with high affinity. The
cell membrane contains a SHBG receptor linked to adenylate cyclase. Activation
of adenylate cyclase by the SHBG receptor or peptide
hormones such as luteinizing hormone generate cAMP, which activates the
protein kinase A (PKA) phosphorylation cascade. PKA can activate the
estrogen receptor-α (ERα) and other steroid hormone receptors
in the absence of ligand binding. Most receptors in the steroid/nuclear
receptor superfamily reside in the nucleus in the newly synthesized
or unliganded state. The exception is the glucocorticoid receptor (GR), which
is complexed with the HSP90 chaperonin complex in the cytoplasm. HSP90 and
its accompanying proteins are thought to stabilize the
receptor until hormone binding occurs. Although receptors for progesterone, estrogens, and
androgens have also been found in complexes with
the primarily cytosolic HSPs, the function of these associations is unclear. In
the case of estrogens, progestins, and androgens, the hormones
freely enter the nucleus and bind to their cognate receptor. Binding
of hormone ligand to the receptor activates the receptor by inducing
alterations in the conformation of the receptor protein. This allows
the receptors to form homodimers and bind to specific hormone response
elements (HREs) (see Table 2) in the DNA in the regulatory region of the target gene. B. Once bound to the HRE, the liganded steroid hormone receptor induces a
DNA bend and, in the presence of agonist ligand, recruits coactivator
proteins. The HRE-bound receptor also interacts with other DNA-bound
transcription factors, as shown for the interaction of Sp1 with ER. Certain
coactivator proteins have histone acetyltransferase (HAT) activity that acetylates lysine residues in histones H3 and H4. This
results in a "loosening" of chromatin structure that facilitates the
binding and transcriptional activity of RNA polymerase II at the transcription
start site. (Adapted from Naar Am, Beaurang PA, Robinson KM
et al: Chromatin, TAFs, and a novel multiprotein coactivator are required
for synergistic activation by Sp1 and SREBP-1a in vitro. Genes Dev 12:3020–3031, 1998; and from Zwijsen RML, Buckle RS, Hijmans
EM et al: Ligand-independent recruitment of steroid receptor coactivators
to estrogen receptor by cyclin D. Genes Dev 12:3488–3498, 1998.)
|
On activation by hormone binding and the release of HSP90 and the other
GR-associated proteins, the hormone-receptor monomer is released from
the complex, dimerizes, and translocates to the nucleus. In contrast
to GR, immunohistochemical localization experiments showed that ERα,69 ERβ,6 PR,70 and AR71 are primarily nuclear in the absence of hormone treatment. All class II
nuclear receptors are nuclear in the absence of ligand.1,4 Although each of the steroid receptors (GR, MR, PR, ERα, AR) has been
found in association with HSP90, with the exception of GR and MR, hormone
binding does not appear to be required for translocation of these
receptors to the nucleus.66 The role of the HSP90 complex in ER function is not clear. Purification
of ERα after chemical cross-linking in intact MCF-7 human breast
cancer cells revealed one ERα monomer complexed with two molecules
of HSP90 and one molecule of p59 (FKBP52) but no HSP70 or 40-kd cyclophilin.72 HSP90 is not required for ligand-dependent transcriptional activation
by ERα.73 Although HSP70 was shown to be required for purified, recombinant ERα to
bind EREs in vitro,74 other experiments imply that HSP70 targets inappropriately folded, artificially
overexpressed nuclear proteins75 and is not required for ERα-ERE binding.76 Entry of Steroid/Nuclear Receptors Into the Nucleus Steroid hormone receptors within living cells are dynamic. They shuttle
between the cytoplasm and the nucleus.66 The hormone receptor enters the nucleus by two processes: passive diffusion
through the an "ever opened" central channel of the nuclear pore
or active transport that is mediated by interaction of the NLSs on the
receptor proteins with the NLS receptor-HSP90 complex.77 The NLS-steroid hormone receptor-NLS receptor-HSP90 complex binds to the
nuclear pore complex by means of nucleoporins in an ATP-dependent process.78 The receptor is then trapped by binding to intranuclear components.77 Steroid hormone receptor complexes have been demonstrated in association
with nuclear membranes and with chromatin components including histones,79 nonhistone basic proteins, DNA, and ribonucleoproteins and with nuclear
matrix.78 The TR, VDR, RAR, RXR, and other class II nuclear receptors do not form
high-molecular-weight complexes but are believed to enter the nucleus
directly and become tightly associated with chromatin.1,4 However, later experiments may cause rethinking of this model. The subcellular
localization of human TRβ1 fused at its N terminus to green
fluorescent protein (GFP) was followed in living cells.80 In the absence of the thyroid hormone T3, more GFP-TRβ1 was present in the cytoplasm, and when the cells were
treated with T3, the GFP-TRβ1 was predominately localized in the nucleus.80 Because GFP-TRβ1 bound T3 and DNA in a manner identical to wild-type TRβ1 and because T3 treatment moved all of the GFP-TRβ1 into the cell nucleus, the investigators
concluded that their findings reflect the behavior of endogenous
TR.80 With the exception of the ERBA1 oncogene product and some of the orphan
receptors that function constitutively or are activated by growth factor-mediated
phosphorylation, unliganded class II nuclear receptors are
not transcriptionally active until liganded, because the class II nuclear
receptors, including RAR, RXR, and TR, are constitutively bound
by corepressor proteins that silence transcription until the appropriate
ligand is bound.32,81,82 Ligand-Dependent Activation of Steroid/Nuclear Receptors The LBD of the receptor may act as a repressor of receptor function because
deletion of the LBD from GRs and PRs causes constitutive gene activation.83,84 Ligand binding to the receptor stimulates dissociation of the receptor-HSP90 complex,2 which facilitates conformational changes in the receptor (activation) that
exposes the DBD and promotes dimerization of the receptor. Binding
of the hormone to the receptor may be only one of several factors that
activates or transforms the receptor, enabling it to bind as a dimer
to specific hormone response elements located adjacent to or sometimes
at a far distance from the transcription start site of the regulated
gene.
Type II nuclear receptors (i.e. TR, VDR, RAR, RXR, and the orphan
receptors) do not interact with HSP90. These receptors are bound to DNA
in the absence of ligand and are associated with corepressor proteins,
such as NCoR81,82
and SMRT.85 Corepressor proteins NCoR and
SMRT are associated with a complex of proteins that have histone deacetylase
activity that are believed to repress gene expression by maintaining chromatin
in a more condensed conformation.86,87,88
Hormone-dependent phosphorylation of steroid hormone receptors may play
an important role in binding of the receptor to its specific response
element on the gene and subsequent activation of transcription. PR, GR, ERα, and
VDR are all phosphorylated after binding to their respective
ligands.89 Ligand-Independent Activation of Steroid Hormone Receptors Although steroid and nuclear receptors are classically activated by ligand
binding and are subsequently phosphorylated, a second mode of activation
in the absence of ligand has been detected for certain receptors. Phosphorylation
of certain steroid hormone/nuclear receptors in response
to cell membrane-activated signaling cascades activates the receptor
in the absence of the cognate ligand.89 Examples of peptide hormones and growth factors that activate steroid
receptors by triggering intracellular phosphorylation cascades include
dopamine, epidermal growth factor (EGF), insulin, and insulin-like growth
factor.89 Activation of β2-adrenergic receptors by the antiinflammatory, antiasthmatic drugs salbutamol
and salmeterol was demonstrated to activate GR, resulting in nuclear
translocation and transactivation of a GRE-driven reporter gene.90 Signal transduction took place through activation of a cAMP cascade mediated
by the cell membrane β2-adrenergic receptor. Likewise activation of the protein kinase A (PKA) pathway
stimulated transcription by the MR in a ligand-independent manner.91 PR appears to be refractory to activation induced by phosphorylation cascades.89 Binding of Steroid/Nuclear Receptors to Hormone Response Elements and DNA
Bending The sequences specifically recognized by the various steroid/nuclear receptors
are described in Table 2. When steroid/nuclear receptors bind their cognate HRE, the DNA is deformed, causing
a bend in the DNA. DNA bending appears to be important
in cellular processes mediated by multiprotein complexes, including transcription
in prokaryotes and eukaryotes92; DNA bending is thought to facilitate interactions between components
of the transcription complex bound to different sites and to promote DNA
looping to allow single proteins to contact multiple DNA elements. ERα binding
to an ERE results in a bend of the DNA toward the major
groove.93 Other steroid receptors, including GR94 and PR95, also induce DNA bending. Similarly, class II nuclear receptors, including
TR and RXR, induce DNA bending.96 That the topology of DNA is important for steroid hormone receptor recognition
of HREs is reinforced by the observation that the nonhistone chromosomal
protein HMG-1, which recognizes irregular DNA structure, enhances
the binding of PR, ERα, AR, and GR to their respective response
elements in vitro but has no effect on the binding of TR, RAR, RXR, or VDR to their target
sequences.95,97 Moreover, coexpression of HMG-1 or HMG-2 increased PR-mediated transcription
in transiently transfected mammalian cells by 7- to 10-fold without
altering the basal promoter activity of target reporter genes.97 Direct Regulation of Gene Transcription by Steroid Hormone Receptors Initiation of transcription is a complex event occurring through the cooperative
interaction of multiple factors at the target gene promoter (Fig. 4). When bound to the specific HRE on the DNA, the hormone-receptor complex
interacts with basal transcription factors and with other proteins
to stabilize basal transcription factor binding and promote the assembly
of the transcription initiation complex. Once the transcription initiation
complex is in place, the enzyme RNA polymerase II is recruited
to the transcription start site, where it begins transcribing the DNA
sequence into mRNA. INTERACTION OF STEROID/NUCLEAR RECEPTORS WITH BASAL TRANSCRIPTION FACTORS. For RNA polymerase II to initiate transcription, basal transcription factors
TFIIA, TFIIB, TFIID, TFIIE, TRIIF, and TFIIH must assemble on the
core promoter.39 TFIID consists of the TATA box binding protein (TBP) and at least eight
tightly associated factors (TAFs) of 18 to 250 kd.98 Because the amount of RNA polymerase II is limited in the nucleus, genes
must compete for it by assembling an appropriate set of cis-acting elements, HREs, and binding sites for sequence-specific transcription
factors, such as AP-199 and Sp1.100 The net result is the synthesis of new mRNAs that move into the cytoplasm
and are translated into new proteins that may alter cell function (acting
in an intracrine manner) or may be modified further and secreted
by the cell to act as endocrine, autocrine, or paracrine factors. Steroid receptors interact with basal transcription factors TFIIB, TBP, and
various TAFs of TFIID.101 Studies indicate that steroid and nuclear receptors use different domains
for interaction with basal transcription factors.101 These interactions help stabilize the assembly of the RNA polymerase II
preinitiation complex. INTERACTION OF STEROID/NUCLEAR RECEPTORS WITH COACTIVATORS. Steroid hormone receptors interact with multiple proteins when bound to
DNA or in solution in vitro. Early experiments showed that overexpression of one type of steroid hormone
receptor could inhibit or "squelch" the activation of transcription
mediated by a different steroid hormone receptor, hinting that steroid
receptors compete for limited amounts of a factor required for transcription.102 During the past 4 years, at least 15 different coactivators have been
identified. Examples of coactivators are listed in Table 3. These proteins have also been called receptor-interacting proteins (RIPs) and
receptor-associated proteins (RAPs); however, not all RIPs are
coactivators. By definition, coactivators are considered to interact
directly with the steroid/nuclear receptor and enhance transcription.32 The first coactivators for steroid receptors to be discovered, the yeast
SWI/SNF proteins,103 may not be strictly coactivators but instead may serve as "bridging factors" that
interact between coactivators and basal transcription factors. Several
coactivators have general transcriptional activator function (e.g. CBP); they enhance transcription by different types of transcription factors, including
steroid receptors. TABLE 3. Coactivator Proteins That Interact With Steroid Hormone Receptors
Names | Interacts With SN/NR | Effect of SR Ligand on Direct Interaction | Effect of Coexpression on Transcription | Other Information | Reference |
ACTR/SRC-3/ | ERα | Requires agonist Ligand | Stimulates E2-dependent transcription | · AIB1 expression elevated in human breast and ovarian cancers238 | 239 |
RAC3/p/CIP/AIB1 | RXRα | | | | 240 |
| | TR | | | | |
ARA70 (ELE1α) | ER | Androgens and antiandrogens promote AR-ARA70 interaction--also genistein and RU486241 | Stimulates AR transcription with DHT or E2 | · no intrinsic HAT activity | 193, 242, 243 |
Truncated | ERα | | | · interacts with p/CAF that has HAT activity | |
variant ELE1β | GR | | | · interacts with TFIIB | |
| PPAR | | | · highest ELE1α and ERE1β expression in testis | |
CBP/p300/p270 | AR, ERα, GR, PPARγ, RAR, RXR, TR, HNF4 | Requires agonist ligand, except for AR | · stimulates transcription | · intrinsic HAT activity244 · CBP/p300 is also a cofactor for AP-1, c-myb, STAT1, E1A, p53, and
Myo-D reviewed in245 | 246–248 |
RIP140 | ERα, TR, RXR, PPARα, PPARγ | Requires agonist ligand; antagonists tamoxifen and ICI 164,384 block interaction
with ERα | · stimulates ERα-induced transcription · inhibits PPAR and RXR activities | · identical to ERAP140249 | 250 |
SRC-1/NoA-1 | AR, ERα, ERβ1, PR, GR, TR, RARβ, RXRα, PPARγ, HNR4 | Requires agonist ligand; antagonist inhibits interaction | Stimulates PR, GR, and ERα induced transcription | Identical to ERAP160 and p160; intrinsic HAT activity107;interacts with p300/CBP, TBP, and TFIIB252,252 | 131 28 |
SWI/SNF | ER, GR RAR, HNF-4 | | Stimulates transcription | Chromatin remodeling complex in yeast with human homologues | 32 |
TIF1α | ERα, ERβ, PR, RXR, VDR | Requires agonist ligand | Stimulates transcription- requires agonist ligand for ERα, but for
ERβ 4-OHT acts as an agonist | Interacts with heterochromatin proteins, including hSNF2b of the SWI/SNF
complex and TIF1β | 132, 253, 254 |
TIF2/GRIP1/NCoA-2 | ERα, GR, AR, PR, RAR, RXR, TR, VDR HNF4 | Requires agonist ligand | Stimulates ERα, AR, PR, TR, RAR, and RXR but not GR, or VDR | 40% sequence homology to SRC-1 | 255 |
These proteins have been identified in yeast and mammalian two hybrid screening, during
purification, in immunoprecipitation assays, and by cross-linking
studies.32,41,101 Some, but not all, have been shown to stimulate transcription in cell
systems.
Steroid receptors can interact with a number of different coactivators. Some
of these coactivators, such as SRC-1, CBP, and TIF2, play a critical
role in ligand-activated transcription.32,101 Coactivator proteins contain one or more copies of a nuclear receptor
binding motif, also called the nuclear receptor box, consisting of the
amino acids LXXLL, which physically interacts with the steroid/nuclear
receptors. Steroid receptors show different affinities for the various
coactivators104 and use different amino acids to contact the coactivators.105 The discovery that coactivators SRC-1, ACTF, and CREB/p300/CBP106 have histone acetyltransferase (HAT) activity107 indicates a possible mechanism for enhanced transcription. Many transcriptional
regulatory proteins have intrinsic HAT activity.108 HATs acetylate lysine residues on the N-terminal tails of histones H3 and
H4 in chromatin, resulting in a weaker association of histones with
DNA, altering nucleosomal conformation and stability in a manner that
facilitates transcriptional activation by RNA polymerase II.109 SRC-1, CBP/p300, CREB, and other coactivators are believed to form a ternary
complex with liganded steroid receptors to increase the rate of
hormone-responsive gene transcription.110 Several HAT activities may be tethered to hormone-activated receptors
on the promoter, yielding synergistic transactivation. Not all genes are
affected by histone acetylation,108 and steroid and nuclear receptors show different affinities of interaction
with coactivators.111 The currently accepted model suggests that different target cells express
different levels of coactivators and corepressors that, along with
the amount of receptor protein and ligand, could allow fine tuning of
target gene transcription in response to steroid hormones. Northern blot
analysis confirmed the idea that different rat tissues112 and cell lines113 express different amounts of the mRNA for p300, CBP, SRC-1, RIP140, SMRT, and
NCoR. Indirect Regulation of Gene Transcription by Interaction of Steroid/Nuclear
Receptors With Other Transcription Factors In addition to their direct action on target gene expression mediated by
direct DNA binding, steroid receptors interact physically with different
transcription factors to alter target gene transcription without
the steroid receptor interacting directly with DNA. The best studied example
of this "transcriptional crosstalk" is the interaction of the AP-1 transcription
factor with GR.114 Depending on the cell type115 and the composition of the AP-1 complex, GR synergizes with AP-1 (JUN-JUN
homodimer) or suppresses the FOS-JUN heterodimer.116 Similarly, ERα liganded by the antiestrogen tamoxifen activates promoters
through AP-1 sites in genes such as that encoding human collagenase.117 This contrasts with the inability of tamoxifen to activate transcription
from promoters bearing classic EREs. Tamoxifen agonism at AP-1 sites
is cell type specific; it occurs in cell lines of uterine but not of
breast origin. The DBD of ERα is required for tamoxifen activation
at AP-1 sites. Conversely, the AP-1 components JUN- and FOS-inhibited, E2-dependent, ERα-stimulated reporter gene activity in transiently transfected
MCF-7 or CV-1 cells transfected with ERα.118 DNA binding experiments revealed that ERα-ERE binding was inhibited
by the JUN protein and that ERαinhibited JUN-DNA binding.118 E2 signals in opposite ways through ERα and ERβ from an AP-1 site: with
ERα, E2 activated transcription, whereas with ERβ, E2 inhibited transcription.119 Moreover, in contrast to ERα, the antiestrogens tamoxifen, raloxifene, and
ICI 164,384 were potent transcriptional activators with ERβ at
an AP-1 site. The two ERs differ in how they respond to ligand
and response element, suggesting that ERα and ERβ may play different
roles in gene regulation.119 Another transcription factor with which ERα interacts directly to activate
gene transcription is Sp1. A number of estrogen-responsive genes
are activated by ERα-Sp1 interaction, including cathepsin D,120 RARα,121 and FOS.122 ERα and Sp1 physically interact and this interaction increases Sp1-DNA
binding in the presence or absence of E2. In contrast, transactivation of Sp1-driven promoter-reporter constructs
is E2 dependent.123 These results indicate that transcriptional activation requires more than
ERα-Sp1 interaction and increased DNA binding. A likely interpretation
is that coactivators are required. Repression of Target Gene Transcription by Steroid/Nuclear Receptors Binding of the hormone-receptor complex can also repress transcription
of target genes.32 One mechanism by which a steroid receptor represses gene transcription
is by binding to the same, or an overlapping, DNA binding site as that
used by a different activator protein, competitively blocking activator
binding to DNA. One example of this mechanism is the mutual inhibition
by GR and AP-1 on the proliferin gene promoter that contains a composite
GRE-AP-1 binding site.116 Competition for limiting amounts of coactivators is another mechanism by
which steroid hormones inhibit gene expression. The mutual inhibition
of AP-1 and steroid receptor transactivation, including AR, ERα, GR, and
PR,114 is achieved through competition for limiting amounts of the coactivators
CBP/p300 in the nucleus.124 Likewise, GR and ERα block NF-κB-mediated transactivation.114 GR and other steroid receptors interact directly with NF-κB, and
NF-κB inhibits GR-, ERα-, and PR-mediated transcription. Later
experiments indicate that GR and NF-κB compete for limited amounts
of the coactivators CBP and SRC-1.125 Another mechanism for repression by steroid receptors is by binding to
the HRE and recruiting corepressor proteins that quench the activity of
activators bound to the promoter. The corepressors N-CoR81,82 and SMRT85 were first identified by their binding to unliganded TR, RAR, and RXR (Table 4). Tethering of these corepressors to the DNA by their interaction with
the DNA-bound TR, RAR, or RXR recruits other components of the corepressor
complex that mediate the actual events of transcriptional silencing.126 Binding of TR or RAR ligands cause dissociation of NCoR, but only when
the receptors are bound to DNA.82 Unlike coactivator complexes that have endogenous HAT activity, corepressors
to not appear to have enzymatic activity. Corepressors NCoR and
SMRT, appear to function by recruiting histone deacetylase (HDAC)-con-taining
multiprotein repression complexes to the promoter.127 Deacetylation of chromatin helps maintain a condensed state of nucleosomal
structure that blocks the binding of components of the RNA polymerase
II transcription initiation complex. TABLE 4. Corepressor Proteins That Interact With Steroid/Nuclear Receptors
Name | Interacts With | Effect of Coexpression on Transcription | Other Information | References |
NCoR | TR RAR RXR PR ERα | Increased transcriptional activity of unliganded PR 256 | NCoR levels were reduced in many breast tumors that had acquired resistance
to the antiproliferative effects of tamoxifen134 | 21, 82 134 20 |
| | | At high levels, NCoR increased RAR transcription257 | | |
SMRT | TR RXR RAR ERα | Increased transcriptional activity of unliganded PR256 | HDAC1 is associated with SMRT | 85 256 |
| | | Overexpression strongly reduced basal and 4-OHT-stimulated gene expression
with no effect on E2 activity133 | | |
These proteins have been identified in yeast and mammalian two hybrid screening, during
purification, in immunoprecipitation assays, and by cross-linking
studies.32,41,101 In general, interaction of steroid and nuclear receptors with these proteins
represses gene transcription in cell systems.
Mechanism of Repression of Target Gene Transcription Achieved by Hormone
Antagonists A variety of mechanisms are likely to be involved in hormone antagonist
action. First, the antagonist activity of an antihormone may depend on
the cell or tissue type. For instance, antiestrogen-liganded ERα binds
EREs with high affinity,50,128 but in certain cell types, such as MCF-7 human breast cancer cells, transcription
is not activated,129 whereas in endometrial cells, tamoxifen stimulates transcription.130 The proposed mechanism for these observations involves the inability of
antiestrogen-liganded ERα to interact with coactivator proteins, such
as SRC-1131 and TIF1a.132 Another possibility is that antiestrogen-liganded ERα may recruit
corepressor proteins to the promoter, inhibiting chromatin changes mediated
by HDACs that promote a "loosening" of nucleosomal structure.133 The corepressor NCoR interacts with the tamoxifen metabolite 4-hydroxytamoxifen (4-OHT)-liganded
ER.134 Breast cancer cells that are resistant to tamoxifen inhibition of cell
proliferation show reduced NCoR levels compared with tamoxifen-sensitive
cells, suggesting a mechanism whereby cells become resistant to tamoxifen.134 Similarly, NCoR and SMRT repress the agonist activity of antiprogestin
RU486-liganded PR or tamoxifen-liganded ERα.21 Nonnuclear Steroid Hormone Receptors and Nongenomic Effects of Steroid
Hormones Although most of the effects of steroid hormones are mediated through their
interaction with their cognate receptors and subsequent effects on
target gene transcription, certain rapid effects of steroid hormones
are incompatible with a transcriptional mechanism.135 Although highly controversial, specific binding sites for estrogen, glucocorticoids, and
vitamin D receptors have been reported in the plasma
membrane of various target cell types.136,137 Estrogen receptors have been reported in the plasma membrane of GH3 pituitary
cells.138 Exposure of GH3 pituitary cells to estradiol was reported to elicit a
rapid (within 5 minute) release of prolactin in a manner that cannot be
accounted for by genomic effects of estradiol mediated through nuclear
ERs. Binding sites for estrogen with different biochemical properties
from the classic nuclear receptor have been reported in the endoplasmic
reticulum of uterine tissues.139 Estradiol appears to have receptor-mediated (genomic) and nongenomic effects
in the brain.140 The cardiovascular protective effects of estradiol are thought to be mediated
at least in part by nongenomic ER and involve increased intracellular
cAMP,141 inhibition of Ca+ 2 influx,142 and stimulation of nitric oxide release.143 Estradiol has exerts direct beneficial effects on human oocytes during in vitro maturation, and these effects at least partly result from steroid action
at the oocyte surface.144 A study compared nuclear and membrane expression of ERα and ERβ by
expressing the cDNAs for each receptor type in Chinese hamster ovary (CHO) cells.145 ERα and ERβ were expressed in the nucleus and in the cell membrane. However, the
membrane receptor number was only 2% of the nuclear
receptors. E2 bound to the nuclear and membrane forms of each isoform with identical
affinity, but E2 bound ERβ with lower affinity than ERα.145 E2 binding to CHO-ERα- or CHO-ERβ-activated G-coupled membrane receptors
Gαq and Gαs and rapidly (within 5 to 10 minutes) stimulated
corresponding increases in inositol phosphate and cAMP.145 Membrane ERα and ERβ showed distinct differences in their activities: JUN
N-terminal kinase activity was stimulated by E2 in ERβ-expressing CHO but was inhibited in CHO-ERα cells.145 Further studies are needed to extend these results on the role of a membrane
ER to human tissues. GRs and MRs appear to have nongenomic effects. GRs bind to cytoskeletal
structures,146 and glucocorticoids stimulate the rapid onset of polymerization of actin
in a nongenomic manner that involves decreased intracellular cAMP.147 GRs may activate mitochondrial gene expression in hepatic cells.148 Rapid stress-induced changes in male amphibian reproductive behavior appear
to be mediated by MRs on neuronal membranes.149 One of the membrane steroid receptors that has been well characterized
is the membrane PR in amphibian oocytes and in spermatids.150 This receptor differs from the nuclear PR. The novel membrane PR mediates
rapid changes in Ca+ 2 conductance in human sperm plasma membrane.151 A high-affinity progesterone-binding membrane protein of 200 kd has been
described in pig liver.152 In addition to membrane PR action, metabolites of progesterone and deoxycorticosterone
act as positive allosteric modulators of the γ-aminobutyric
acid (GABA)A receptor complex in the cortex of rat brain.153 This means that these hormones bind to an effector site (not the GABA
binding site) and increase the affinity of GABA binding to the GABAA receptor. Progesterone also binds to the G-coupled oxytocin receptor and
inhibits peptide-mediated signaling.154 The role for these steroid membrane receptor effects in human health is
just beginning to be explored. |