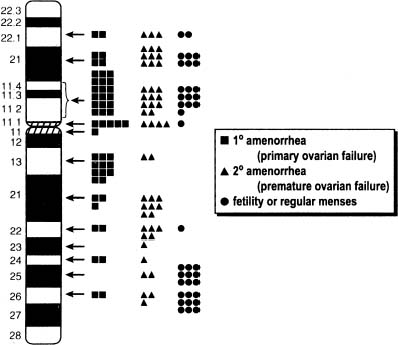
Historically, the term applied to women with ovarian failure is gonadal dysgenesis or Turner syndrome. Turner syndrome is broad term, however, and here the term gonadal dysgenesis is applied to women with streak gonads and use the term Turner stigmata for those having short stature and certain somatic anomalies (Table 1). By itself, Turner stigmata as defined would not imply the presence of streak gonads. The term Turner syndrome would be applied to those individual with both streak gonads, Turner stigmata and a 45,X or X-deletion complement.
TABLE 1. Somatic Features Associated with the 45,X Chromosomal Complement
Growth
Decreased birth weight
Decreased adult height (141 to 146 cm)
Intellectual function
Verbal IQ higher than performance IQ
Cognitive deficits (space-form blindness)
Immature personality, probably secondary to short stature
Cranofacial
Premature fusion sphenoccipital and other sutures, producing brachycephaly
Abnormal pinnae
Retruded mandible
Ptosis
Hypertelorism
Epicanthal folds (25%)
High-arched palate (36%)
Abnormal dentition
Visual anomalies, usually strabismus (22%), anbtyopia
Hypernetropia auditory deficits; sensorineural or secondary to middle ear
infection
Neck
Pterygium colli (46%)
Short, broad neck (74%)
Low nuchal hair (71%)
Chest
Rectangular contour (shield chest) (35%)
Apparent widely spaced nipples
Tapered lateral ends of clavicle
Cardiovascular
Coarctation of aorta or ventricular septal defect (10% to 16%)
Renal (38%)
Horseshoe kidneys
Unilateral renal aplasia
Duplication ureters
Gastrointestinal
Telangiectasias
Skin and lymphatics
Pigmented nevi (63%)
Lymphedema (38%), generalized, caused by hypoplasia superficial
vessels; puffy hands and feet
Nails
Hypoplasia and malformation (66%)
Skeletal
Cubitus valgus (54%)
Radial tilt of articular surface of trochlear
Clinodactyly V
Short metacarpals, usually IV (48%)
Decreased carpal arch (mean angle 117degree)
Deformities of medical tibial condyle
Dermatoglyphics
Increased total digital ridge count
Increased distance between palmar triradii a and b
Distal axial triradius in position t
Percentage affected reflect tabulation of Simpson, from which table is modified.22
Cytologic Origin
In 80% of 45,X cases the X is maternal (Xm) in origin. In the remaining 20% the remaining X is paternal (Xp) in origin.9,10
Monosomy X
GONADS.
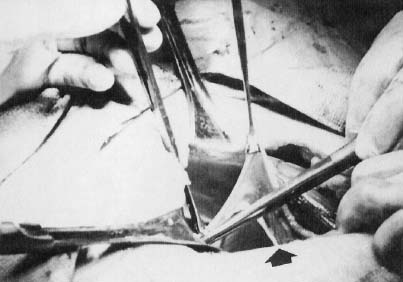
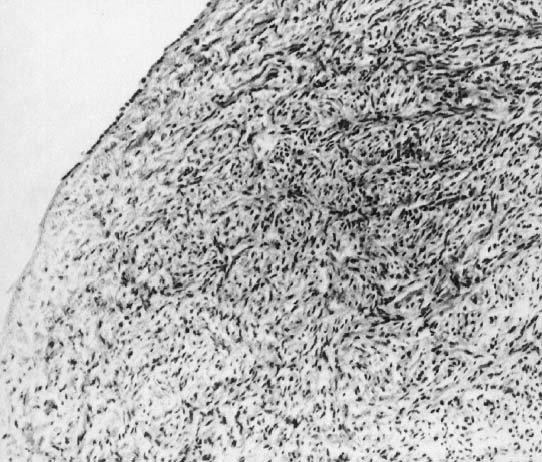
In monosomy X the gonad usually exists not as the typical ovoid structure but as a white fibrosis streak, 2 to 3 cm long and approximately 0.5 cm wide, located in the position ordinarily occupied by the ovary (Fig. 1). A streak gonad is characterized histologically by interfacing waves of dense fibrosis stroma (Fig. 2).
|
|
That 45,X individuals show streak gonads as adults is not as obvious as might be expected, given that relatively normal ovarian development occurs in many other mammals (e.g., mice) with monosomy X. The presumptive explanation is that pivotal genes on the normal heterochromatic (inactive) X are being inactivated. Of the some 2000 genes on the X, only perhaps 5% escape inactivation.11 Most of these genes are on the X short arm (Xp), clustered in selected euchromatic regions. Candidate genes for ovarian maintenance genes will probably prove to lie in these euchromatic regions.
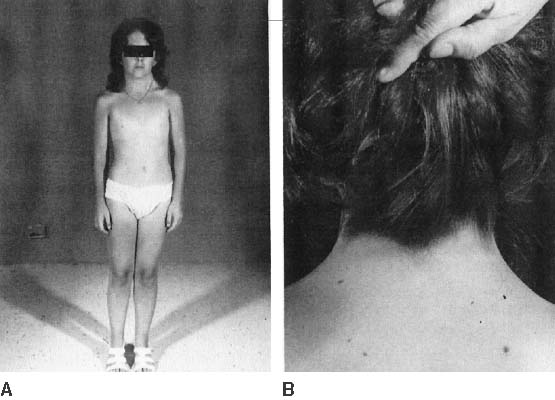
The endocrinologic correlates of ovarian failure are deficient secretion of sex steroids. Estrogen and androgen levels are thus decreased; follicle-stimulating hormone (FSH) and luteinizing hormone (LH) levels are compensatively increased. Deficiencies of estrogen-dependent processes leads to predictable effects of hormonal deficiency. Premenarchal uterine enlargement and growth spurt are not observed. Pubic and axillary hair fail to develop at puberty (Fig. 3). Breasts contain little parenchymal tissue, and areolar tissue is only slightly darker than the surrounding skin. External genitalia, vagina, and müllerian derivates are well differentiated, but remain small (unstimulated) in the absence of exogenous steroids.
|
Approximately 3% to 5% of adult 45,X patients menstruate spontaneously (at least twice) and show breast development. Many fertile patients have been reported, as recently reviewed by Abir and colleagues12 and Hovatta.13 An undetected 46,XX cell line (i.e., 45,X/46,XX mosaicism) should be suspected in menstruating 45,X patients. This is especially plausible in reports like that of Magee and colleagues14 who observed seven pregnancies in one ostensibly 45,X woman. However, it is not expected that some 45,X individuals could be fertile, inasmuch as germ cells are present in 45,X embryos. In addition, pregnancy can occasionally be achieved in hypergonadotropic women by sequential gonadotropin suppression followed by ovulation induction. Check and colleagues15 induced ovulation in 5% of 361 cycles in 100 hypergonadotropic women; the chromosomal complements of women were not stated.
Hormonal treatment of 45,X women usually involves hormone therapy (estrogen and cyclic progestogens). This can result in normal uterine size, which could be followed by assisted reproductive technology (ART) if pregnancy is desired. The process involves a partner’s sperm being mixed with ova donated from another woman, fertilization in vitro, and transfer of embryo to the uterus of the hormonally synchronized 45,X patient. The success rate (clinical pregnancies) is over 20% per cycle. Thus, Foudila and colleagues16 reported 20 clinical pregnancies among 18 women with Turner’s syndrome, the exact chromosomal complements not being stated. Although the clinical pregnancy rate per fresh embryo transferred was impressive at 46% (13/28), 7 of the 13 (54%) resulted in spontaneous abortion, for a take-home baby rate of 46% (6/13). Among transferred frozen embryos the rates were 28% (7/25) and 14% (1/7), respectively. Low pregnancy rates were also reported by Khastgir and colleagues17 and Tarani and colleagues.18 The ability of a 45,X woman to carry her pregnancy to term must be addressed before embarking on ART. Specifically, women with coaratation of the aorta may be unsuitable candidates. The increased prevalence of autoimmune disease (e.g., thyroiditis), carbohydrate dysfunction (diabetes mellitus), hypertension, and other adult-onset diseases further places many 45,X patients in high-risk situations.
In the opinion of the author, offspring of 45,X women show little, if any, increased risk for chromosomal abnormalities.19,20 Claims to the contrary12 need to be tempered by biases of biases of ascertainment not being taken into account. Outcomes are relatively normal in pregnancies observed after the index case was diagnosed (truncate analysis). Adverse outcomes prior to that time were probably the reason cytogenetic studies were initiated. Spontaneous abortions are increased donor oocyte ART. This probably reflects hormonal dysfunction or uterine factors (hypoplasia), rather than transmission of aneuploid (monosomic) gametes.
Somatic Anomalies
45,X individuals not only are short (less than 4 feet 10 inches) but often exhibit various somatic anomalies (Turner stigmata) (Table 1). No single feature(s) of Turner stigmata is pathognomonic, but in aggregate a characteristic spectrum exists that is more likely to exist in 45,X individuals than in individuals having most other sex chromosomal abnormalities. Systematic evaluation of renal, vertebral, cardiac, and auditory function is obligatory, irrespective of the patient’s age when diagnosed.
Growth
45,X neonates are usually low birth weight. Total body length at birth is less than normal, but often close to the 50th percentile. Before puberty, height velocity falls in the 10th to 15th percentile,21 and the mean height of untreated 45,X adults (younger than 16 years old) is between 141 and 146 cm,22,23 perhaps 20 cm less than normal. In normal females the predicted adult height can be estimated by summing the heights of both parents, dividing by 2, and subtracting by 13 cm.24 Taking into account decreased expected height for 45,X individuals, correlation of the height of a 45,X offspring with midparental height holds for Turner syndrome as it does for normal 46,XX females.25 That is, absolute height predicted in Turner syndrome is less but the midparental height correlation still holds.
Various treatments for short stature in 45,X patients have proposed, including growth hormone (GH), anabolic steroids and low-dose estrogen.26 Most treatment regimens show ostensible benefit, especially immediately after onset of therapy. Consensus is now that that ultimate height can be increased by 6 to 8 cm by GH treatment alone.27,28 The most popular form of treatment is human recombinant DNA-derived human GH. GH results in an 8.4 ± 4.5 cm increases in height over that predicted; final height was 150.4 ± 5.5 cm in one heterogeneous group.29 With growth hormone and oxandrolone, the increase was 10.3 ± 4.7 cm. Treatment regimens are discussed in standard pediatric endocrinologic treatsies (e.g., Grumbach and Conte 30). In general, treatment is begun at 2 to 5 years of age, and stopped at approximately 15 years of age.31 Low-dose estrogen is deferred until final height is near29; high-dose estrogen should then be given to stimulate secondary sexual development. The latter regime should begin at 14 to 15 years of age, starting with 0.3 to 0.625 mg conjugated equine estrogens daily for 6 to 12 months; dose is then increased to 1.25 mg.
A potential reason for limited efficacy of growth hormone treatment may be that epiphyses in 45,X individuals are structurally abnormal. Not only long bones, but teeth32 and skull33 are also abnormal. Thus, patients with a 45,X chromosomal complement could be said to have a skeletal dysplasia.
Some wonder whether presence of an unappreciated Y-bearing line (e.g., 45,X/46,XY) could lead to neoplastic consequences with treatment. However, few ostensibly nonmosaic 45,X cases will prove to have a 46,XY even after analysis of thousands of cells using FISH with a Y probe.
Intelligence
Most 45,X patients are of normal intelligence, but any given patient has a higher probability of being retarded than a 46,XX person.22 Performance IQ is lower than verbal IQ, the latter being similar to 46,XX matched controls. 45,X individuals may have a cognitive defect characterized by poor spatial processing skills (space-form blindness). Ross and colleagues34 opined that only loss of distal Xp (Xp 22.33) produced this phenotype; regions responsible for neurocognitive deficits were believed distant from statural or ovarian abnormalities.
Psychosocial deficits primarily reflect behavioral immaturity and difficulties in social relationships. These are probably secondary to delayed sexual development and statural growth.35,36 The possibility has been raised that parental origin influences phenotype Xm (X of maternal origin). Xm cases are said to show cognitive deficits more often than Xp.37 If true, this would indicate the X contains imprinted genes.
Adult-Onset Diseases
Many adult-onset disorders occur in 45,X cases with frequencies greater than expected in the general population. Hypertension deserves special comment, given its presence in about one third of adults 45,X individuals. Hypertension need not alter hormonal therapy; however, careful monitoring is required, and exogenous estrogen therapy may need to be reduced. Frequencies of diabetes mellitus and autoimmune thyroiditis are increased.
Is a Particular Region of the X Responsible for Somatic Anomalies in 45,X?
The region of the X responsible that if deleted results in somatic anomalies is unclear, but it is not necessarily the same as that for ovarian maintenance. The distal X short arm (Xp) has in particular been implicated in somatic development. A pseudoautosomal gene implicated in somatic development and short stature (Turner stigmata) is SHOX. This pseudoautosomal locus is not subject to inactivation. RPS4X is another candidate for the same reason. Zinn and colleagues38 and Zinn and Ross39 have attempted to correlate somatic anomalies with Xp perturbations, using molecular markers to define deletions and X/autosomal translocation. High-arched palate, short stature, and autoimmune thyroid disease were associated with terminal deletions of Xp11.2-22.1, the same region noted to contain ovarian determinants. Boucher and colleagues40 concluded that Xp11.4 is critical for lymphoedema. Bioné and Toniolo41 have also discussed candidate genes on Xp and Xq that could be important for somatic differentiation.