
Inflammatory and Vascular Placental Pathology
Authors
INTRODUCTION
The role of the placental pathologist in clinical obstetrics and neonatology has long been controversial. Obstetric endorsement of the utility of placental histologic examination is commonly lukewarm, especially from obstetricians who do not have a placental pathologist as part of their own local clinical care team. Placental pathologic examinations are pointless if they do not provide clinically useful data. For that reason, we have directed our efforts toward clarifying the contributions and limitations of placental examination to clinical practice. One important limitation is the inability to make a one-to-one link between any one placental or uteroplacental vascular lesion and a particular maternal or fetal/neonatal problem. This would be an unrealistic goal, because in no other organs are such relationships the rule. Neither hepatocellular necrosis nor glomerulosclerosis, for example, is diagnostic of one and only one hepatic or renal disease; in those diseases, individual lesions are considered as part of a greater histopathologic pattern. Finally, the pattern is correlated with a variety of clinical data, laboratory data, and additional pathologic studies to produce the final clinical/pathologic diagnosis. This comprehensive approach yields not only a summary “label” for the histopathologic findings but also information regarding etiology, prognosis, and optimal therapy. It is illogical to expect that the placenta and its diseases should be any more or less complicated than diseases of the liver and kidney. Obstetric technologies and therapies are also directed toward general pathophysiologic processes (such as uterine and uteroplacental Doppler velocimetry, anticoagulant therapy, and maternal immunization and intravenous gamma globulin) rather than specific lesions or tissue diagnoses. Therefore, we are addressing in this chapter the processes of acute inflammation and vascular injury, rather than a series of independent lesions. In factor analyses, where a computer program searches for patterns within a data set, patterns of tissue injury have been found to be specific for, for example, principal indication for extreme preterm delivery (see below). One of us (C.M.S.) is developing data sets to address patterns of tissue injury in preterm birth across ethnic and racial groups. Such an analysis, we hope, may help us to understand the persistent health disparities of African-American newborns in the United States.
The principal goal of this chapter is to provide a conceptual framework for the obstetrician to better understand the answer to the following questions: What is the underlying process that initiated placental injury, and when in gestation did it begin? What secondary processes may have been recruited (and when) to contribute to the final pathology and pathophysiology of the given pregnancy?
ACUTE INFLAMMATORY PLACENTAL DISEASE
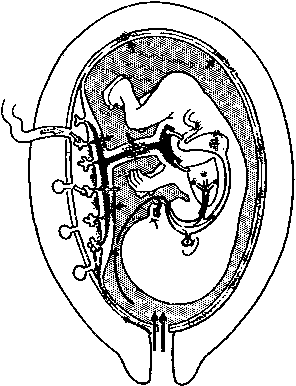
Intrauterine infection can follow ascending, hematogenous, transabdominal, or transfallopian pathways. The most common route is ascent from the perineum, cervix, and vagina (Fig. 1). The causative organisms may be of low virulence, part of these regions' normal flora, or obvious pathogens (e.g., Escherichia coli). They may also be part of a common pool that is traded between mother and spouse. A recent study showed that 70% of consorts of pregnant women with the “subacute amniotic infection syndrome” had pathogenic bacteria, compared with 30% of semen in the control group.1 Further, the perinatal loss rate was 6.7% in the group that was successfully treated and 55% in the group with persistent infection despite therapy.1 This study may point to the role of the male reservoir of potential pathogenic bacteria, but the finding of 30% positivity of controls also highlights the contribution of individual host immunity and local, possibly mechanical factors in the actual ascent of pathogens.
|
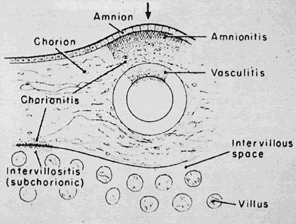
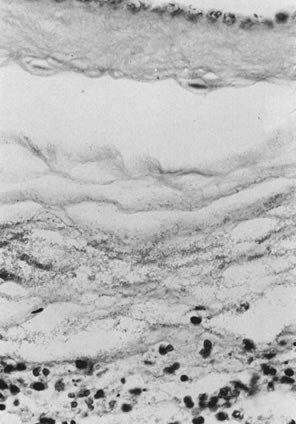
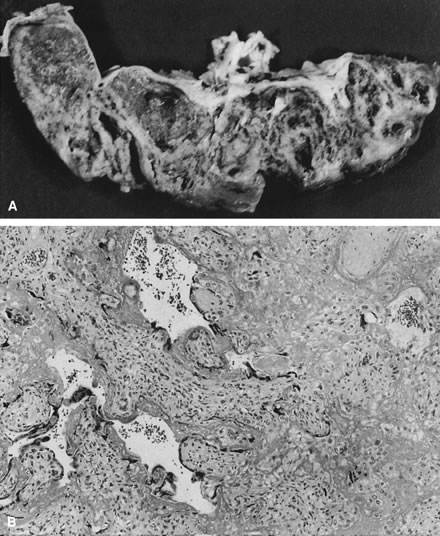
The infectious agent may colonize the extraplacental membranes or may cross intact or ruptured membranes into the amniotic fluid. If there is decidual or choriodecidual colonization, neutrophils are recruited from the decidual vasculature (by chemotactic cytokine stimuli generated locally). These cells may stay local to the chorion and decidua (rather than migrating out of these tissues toward the amnion/amniotic fluid space). In this pattern of inflammation, termed necrotizing,2 tissue destruction and neutrophil infiltration of the decidua and chorion predominate, with pockets of neutrophil debris studding the residual decidua. On the other hand, bacteria or their toxins may enter the amniotic fluid proper. Then, intra-amniotic cytokines call neutrophils out of the decidua, across the chorion, and into the amnion, often leaving the decidua and chorion well preserved, with little nuclear debris or damage. The fetus also responds with recruitment of inflammatory cells out of the fetal blood flow (Fig. 2).2 “Marginating” inflammation predominates if amniotic fluid colonization is an early part of the infectious process (Fig. 3). A combined pattern of both destructive or necrotizing deciduitis and a margination toward the amnion can be seen when amniotic fluid inflammation occurs comparatively later in the infectious process. Bacteria or their toxins in the amniotic fluid elicit maternal neutrophil margination first in the subchorionic space. Recruited neutrophils then migrate across the chorion and amnion into the amniotic fluid (Fig. 4). Microscopic examination should include samples from the zone of spontaneous rupture of the membranes, the pericervical membranes being the area first affected when infectious agents ascend across the cervix. The extraplacental membranes distant from the site of rupture (often a membrane roll), sections of umbilical cord, and the chorionic plate are required to provide clues to the following questions: Did bacteria gain access to the amniotic fluid space? Did the fetus respond to the infectious process? It is this fetal response that may be critical to serious long-term sequelae, especially those related to prematurity, including lung disease3 and brain damage.4, 5, 6, 7
|
|
|
The extraplacental membranes most often rupture because they are already infected. However, the loss of the mechanical membrane barrier to ascending organisms and also the loss of the bacteriocidal/static amniotic fluid may facilitate subsequent ascent of additional numbers or types of microorganisms. We found, in preterm deliveries, no increased severity of maternal acute inflammation (in amnion, chorion, decidua, and chorionic plate) with increasing time from rupture of membranes to delivery, although fetal inflammation (in the umbilical cord) tended to be more severe the greater the latency period.8 This may reflect poor sensitivity of the histologic grading system (i.e., failing to detect increasing neutrophil density after the neutrophils become “too numerous to count”) or may be the histologic reflection of the relative immune immaturity of the preterm fetus. The comparatively immune-incompetent fetus may require more time to respond to any given intra-amniotic stimulus with a histologic inflammatory infiltrate.
Acute intra-amniotic infection may be the single identifiable predisposing factor in as many as 50% of premature deliveries, although only 8–25% of mothers are symptomatic.9, 10 The correlation between the clinical and histologic diagnosis of acute ascending infection in preterm gestations is less than 50%, even though infections are generally more severe in preterm gestations. While few infants are septic at the time of delivery,9 the inflamed fetus may show decreased fetal well-being (drop in biophysical profile score), abnormal fetal heart rate, and possibly increased umbilical systolic/diastolic ratio.11, 12, 13, 14
The histologic pattern of acute inflammation in the amnion, umbilical cord, chorionic plate, chorion, and decidua does not occur in response to intrauterine fetal demise or tissue staining by sterile meconium. Inflammation in the subchorionic fibrin (margination of maternal cells) is the earliest sign of intra-amniotic colonization; however, in such cases amniotic fluid cultures are rarely positive9 because of the small inoculum or the fastidious nature of the organism. Histologic grading of acute inflammation assesses the density of neutrophil infiltrates and the relative distance migrated from the vessels of origin. Thus, variation in the intra-amniotic bacterial load, bacterial virulence, maternal immune competence, and antibiotic therapy along with the duration of exposure to the infectious agents combine to yield the final picture of histologic inflammation in the delivered placenta. By histologic examination alone, it is not possible to precisely extract the time of onset of infection.
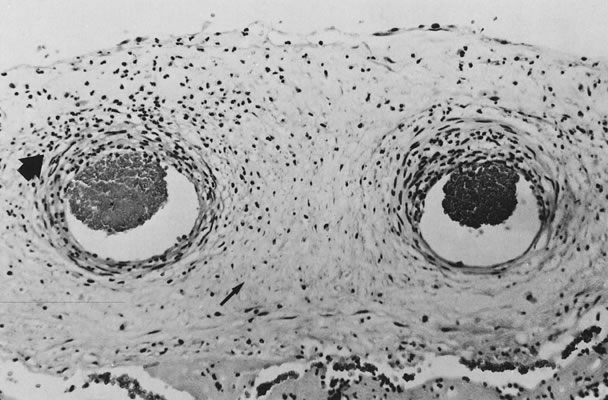
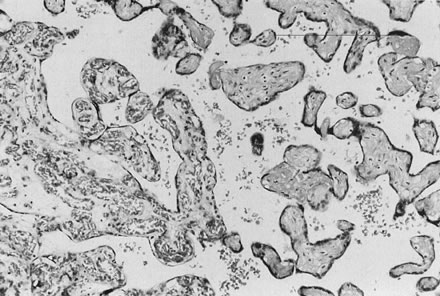
If specimens are cultured for aerobic and anaerobic bacteria in addition to Mycoplasma and Ureaplasma, organisms will be recovered in more than 75% of cases; many are mixed infections. Fusobacterium is an anaerobic filamentous bacterium that results in necrosis of the amnion and is often associated with preterm delivery.15 Group B streptococci may result in “clouds” of bacteria without an appropriate maternal inflammatory response, especially in chronic carrier states (Fig. 5).16, 17 Diverse Candida species may also be pathogens (Fig. 6).18 Gram-negative enteric bacteria were recently isolated from between the chorion and amnion in 55% of preterm deliveries, compared with 26% of term births. There was no significant difference in the patterns of isolation of other organisms by gestational age.19 It is likely that different pathogens may be primary in different populations, and an understanding of the local distribution of flora would be important to population-specific applications of microbial surveillance and antibiosis.
|
|

The fetal response to intra-amniotic infection is shown by marginating fetal leukocytes in the umbilical cord and chorionic vessels. Inflammation commonly but not always begins in the vein. Because the umbilical cord is often thinner at the placental end (close to the chorionic insertion), significant cytokine gradients (and histologic inflammation) may first be established here. However, loops of umbilical cord that are positioned in the lower uterine segment may have a greater exposure to ascending bacteria and may show segmental inflammation. At least two samples of umbilical cord should be examined in any case in which intra-amniotic infection is suspected because of the variability in the histologic pattern. Inflammation is not seen in the intra-abdominal umbilical vein or ductus venosus. Umbilical vasculitis is a more specific predictor of a positive amniotic fluid culture than is maternal inflammation in the chorion, decidua, and chorionic plate.2 Because the fetal white cell pool is relatively small, in severe infections immature neutrophils and even eosinophils may be recruited to umbilical and chorionic vessels from hepatic myelopoietic sites by cytokine stimuli carried by umbilical venous blood. Eosinophils are, in our experience, uncommon in histologically “low-grade” intra-amniotic infection. Umbilical venous cytokine levels parallel the severity of fetal histologic inflammation.20
Eosinophils are occasionally a prominent cell in the fetal infiltrates, especially in preterm infants. This may reflect depletion of the comparatively small preterm myeloid pool, with recruitment of eosinophils as back-up. Circulating eosinophilia is uncommon even in newborns with many eosinophils in their perivascular exudates. Sclerosing (necrotizing) funisitis (Fig. 7) is caused by long-standing intrafunicular infection in which the fetal inflammatory cells have lysed and become calcified within Wharton's jelly. Funisitis is uncommon before 26 weeks because of the relative immunoincompetence of the fetus.21 However, when it is present, it can be severe. For this reason, the relative intensity of fetal inflammation relative to maternal inflammation at any gestational age varies. If the mother is immunoincompetent (e.g., maternal human immunodeficiency virus [HIV] infections), the fetal inflammatory response may be disproportionately greater than expected for the level of maternal inflammation. The same disproportion between fetal and maternal inflammatory responses has been seen in cases of catastrophic fetal group B streptococcal sepsis. Genetically determined variance in cellular and/or humoral immunity to the streptococcal organism may explain differences in fetal and neonatal outcomes in cases of maternal streptococcal colonization.
|

PLACENTAL LESIONS ASSOCIATED WITH ACUTE INFLAMMATION
Placental lesions that may occur more commonly in cases with histologic acute inflammation include villous edema, perivillous fibrin/fibrinoid, placental abruption, and meconium passage. Villous edema must be distinguished from hydrops (Fig. 8). It is especially common in preterm placentas with acute ascending infections, possibly because the looser stroma of the preterm villus facilitates accumulation of extravascular fluid. Although villous edema has been associated with increased perinatal morbidity,22 the confounding factors of prematurity itself, as well as the more severe infections in spontaneous preterm births, may explain this association. We have not seen any association of villous edema with perinatal outcome once we have controlled for the presence and severity of acute inflammation.23 Villous edema per se reflects the effects of any change in trophoblast permeability/integrity, intravascular pressure, and/or capillary integrity and thus is a final common pathway for a wide range of pathophysiology. Fluid leak due to hypoxic damage to capillaries or trophoblasts and changes in intervillous and/or fetoplacental intravascular perfusion pressure are a few of the potential antecedents to villous edema. Villous edema may be more common, and more severe, in the more “acute” stages of infection. When dense neutrophil karyorrhexis in membranes and plate speaks for a long-standing intrauterine infection, villous edema may be minimal. Villous edema in cases with intra-amniotic infection may also be due not to the inflammation but to the hemodynamic perturbation of acute abruption, changing placental intravascular pressure and fluid balance.
|

Given that intrauterine bacteria may serve as an exogenous and proliferating source of uncontrolled stimulation to an unready myometrium, it is not surprising that there is an increased incidence of placental abruption in association with acute ascending infection.24 Neutrophil products may also directly affect decidual integrity; granulocyte elastase release may reduce fibronectin receptor expression in decidual cells, weakening adherence of decidual cells and predisposing to placental separation.25 In light of these findings, the association of abruption with premature membrane rupture (a second locus for pathology related to defective decidual adherence) is of interest.26 In general, the neutrophil infiltrates in extraplacental membranes break off abruptly at the placental margin in acute intra-amniotic infection. It is often striking how sharp the transition between densely inflamed membranes and well-preserved basal plate can be. Acute decidual inflammation is a reflection of tissue injury by any of a number of pathways. Recently, acute basal deciduitis has been suggested to reflect an infectious cause of stillbirth.27 Inflammatory involvement of basal plate decidua was sevenfold more common in stillbirths than in liveborn controls. We find acute basal deciduitis to be very common in abruption, via basal decidual destruction by retroplacental hematoma eliciting an inflammatory response. The observed association of acute basal deciduitis in stillbirth may be explained by stillbirth being more common in cases of acute infection complicated by abruption. Not all acute inflammations in the uterus can be equated to “infection.” In addition to simple tissue injury, acute progesterone withdrawal can cause focal neutrophilic deciduitis in sheep.28
Both perivillous fibrin deposition and villous fibrinoid necrosis are lesions that develop external to at least the trophoblast basement membrane29 and are more common in acute ascending infection. Nelson and colleagues30 have provided evidence that perivillous fibrin/fibrinoid may be an integral part of syncytial repair, especially in cases of syncytial apoptosis. To complicate matters, there may be two very distinct types of perivillous fibrin/fibrinoid, one type essentially a blood clot and the second a “matrix” type that is immunopositive for extracellular matrix molecules such as the fibronectins, collagen IV, laminin, and tenascin.31 Inflammatory cytokines may induce apoptosis in specific cell types32 and also initiate coagulation.33 Intervillous cytokine infusion has been shown to produce lesions of perivillous fibrin/fibrinoid,34 and maternal cytokine infusions have been shown to produce placental necrosis in animal models.35 This investigative line opens a novel mechanism for inflammation-related placental dysfunction—namely, cytokine-associated trophoblast damage.
Finally, the common coincidence of meconium histiocytosis and acute ascending infection has been explained in different ways. Acute ascending infection with intra-amniotic colonization creates the potential for fetal swallowing of bacteria and inflammatory cytokines. This in turn might cause fetal gastroenteritis and diarrhea (and meconium passage).36 If this pathway is the basis for meconium passage, one might expect meconium passage in cases with either a greater load of bacteria or a longer duration of fetal exposure to inflammatory cytokines. Although histologic acute inflammation is generally more severe in preterm cases than at term, most preterm cases do not pass meconium. Meconium passage is uncommon at less than 32 weeks, and if pigment is seen in the membranes or plate in such a case, hemosiderin (a marker of intrauterine bleeding) may be suspected. Prussian blue stains for hemosiderin can differentiate cases with blood breakdown products rather than meconium in the placental tissues, often a clinically relevant distinction.
At term, there are at least three differences that could affect how, when, and why meconium is passed: the closer location of meconium to the anal sphincter at term, making passage easier with less stimulus; the lack of clinical indications to delay delivery at term; and decreased intra-amniotic volume at term, making cord compression events more likely.
The most common process underlying term meconium passage may be transient stresses related to fluctuations in umbilical cord hemodynamics. In this case, acute intra-amniotic infection may only provide vasomotor stimuli (inflammatory mediators) that enhance the umbilical cord's vasoconstrictive response to compression. This response may be exaggerated in the context of umbilical vascular smooth muscle damage caused by cord trauma or, in unusual cases, associated with meconium infiltration of the cord and/or vascular media (Fig. 9). Whether vasospasm is the mechanism by which meconium-associated vessel injury affects the placenta (and fetus) or some other process is invoked is unclear.
|

CHRONIC INFLAMMATORY PLACENTAL DISEASE
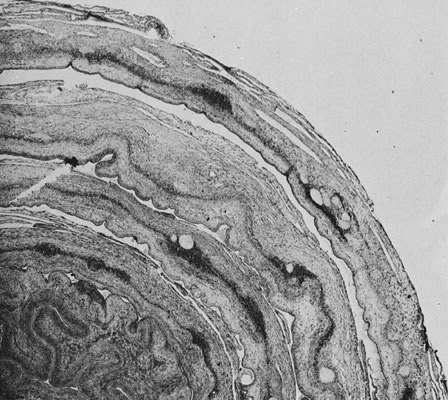
Placental infections via the maternal bloodstream, hematogenous infections, are most often associated with villitis, inflammation of the placental villi. Acute villitis and decidual and villous microabscesses are caused by maternal bacterial sepsis, most commonly with Listeria monocytogenes. Maternal viral infections are more often associated with chronic villitis or chronic choriodeciduitis and/or amnionitis (Fig. 10).37 Chronic chorioamnionitis may also be caused by chronic bacterial infections such as syphilis. In chronic villitis, the inflammatory cell types may differ between infectious agents but unfortunately are not specific for different etiologic agents. Cytomegalovirus also can damage villous endothelium, and affected villi may have variable amounts of hemosiderin, reflecting fetal vascular injury. Granulomatous villitis may be caused by various viruses, mycobacteria, fungus, or parasitic agents. Chronic umbilical vasculitis, including sclerosing funisitis with perivascular calcification, may also be caused by viral and chronic bacterial infections.38, 39, 40 Case reports continue to expand the range of fetal anomalies seen in early ToRCH and related viral infections, including varicella zoster.
|
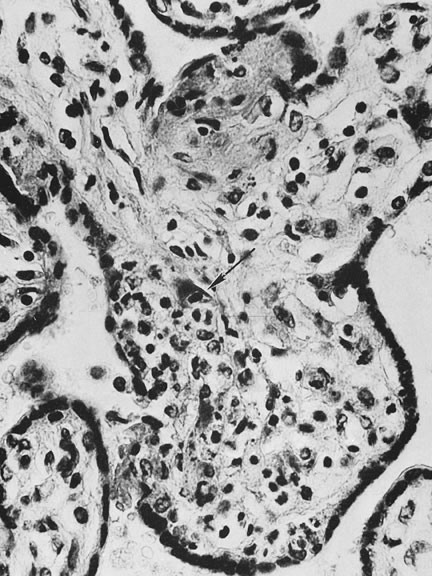
Ultrasonographic identification of the typical patterns of fetal damage can be performed by the midtrimester.41 With organisms such as varicella zoster, the patterns of fetal damage directly implicate the virus. For other viruses, their mode of effect may be more complex and include placental dysfunction via placental cytokine induction.42 Viral infection may selectively damage those placental cell types that are susceptible to infection (i.e., express the viral receptor). For example, the coxsackie and adenovirus receptor may be constitutively expressed in invasive or extravillous trophoblast cells, but not in villous trophoblast. Adenovirus infection, by selectively damaging the invasive trophoblast component, could disturb placental invasion and result in later placental dysfunction.43 A similar pathway (of selective infection of invasive cytotrophoblasts) has also recently been proposed for cytomegalovirus-associated reproductive compromise.44
Finally, bacterial and viral infections may work together to modify placental sensitivity to other infectious agents. Whether the test system involved double virus-infected cultures or bacterial factor-treated cultures, two effects can be observed. In virus-sensitive tissues (those without innate immunity), pretreatment of the tissues with bacterial factors (such as lipopolysaccharide) or double infections inhibited viral replication. In tissues with innate immunity, the same treatments stimulated viral replication. It might be presumed that the placenta is without innate immunity, and that inhibition would be favored in utero. Endogenous tumor necrosis factor-alpha was produced in both stimulation and inhibition reactions.45 Malarial infection enhances expression of the CC-chemokine receptor 5 on placental macrophages. This would increase the number of HIV target cells in the placenta and increase the size of the potential viral reservoir in the placenta.46 Upregulation of this chemokine receptor has been associated with vertical transmission of HIV.47
Placentas from HIV-positive women do not have any specific features. There is a higher rate of acute chorioamnionitis in HIV-infected women, particularly those who have AIDS-defining characteristics.48 Increased transmission of HIV to the infant has also been associated with histologic chorioamnionitis.48, 49, 50 The chorioamnionitis and funisitis appear to be secondary to usual ascending bacterial infections. Rarely, acute or chronic villitis has been reported.51 The ratio of fetal to placental weight may be significantly decreased in HIV-positive pregnancies, with a relative villous hypercellularity51 that may suggest a proliferation of villous stromal monocytes/macrophages. Viral load within the placenta is very low, and routine immunohistochemical techniques for demonstrating p24 antigen and gp41 antigen are consistently negative. In situ hybridization and in situ polymerase chain reaction have been more successful in demonstrating virus within placental tissues.52 The placenta acts as a good barrier to infection, and demonstration of viral antigens in placental tissues does not predict fetal infection. Cells found to contain HIV include syncytiotrophoblasts, cytotrophoblasts, Hofbauer cells, and endothelial cells.53
Chronic villitis may be potentially noninfectious or of undetermined or obscure etiology (“villitis of undetermined etiology,” Fig. 11).54 In some cases, infectious agents may have left no clinical or histologic traces. Chronic villitis without positive viral cultures should be considered as being of undetermined etiology. Then the possibility of the controversial and still overall poorly characterized maternal “immune” pathologies may be raised.55, 56 Chronic villitis is seen in recurrent pregnancy failure without any evidence of recurring infection.55 In our 15-plus years of histopathologic review of patients with recurrent pregnancy loss, chronic inflammatory pathology is a comparatively common finding in patients with clinical autoimmune disease and subclinical (e.g., serologic) evidence of autoimmunity.57 The implications of chronic villitis for future pregnancies depend on intercurrent maternal disease and past pregnancy outcome and may be influenced by family history of immune disease. Chronic villitis is reliably58 diagnosed by the presence of lymphocytic villous stromal infiltrates; the infiltrate may include lymphocytes, monocytes/macrophages, plasma cells, and rarely eosinophils.
|
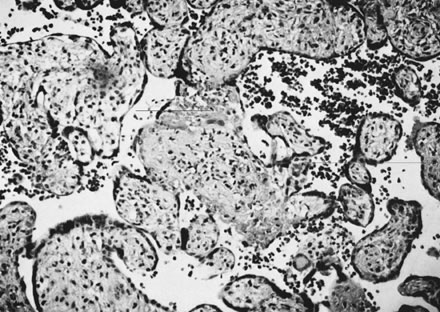
The diagnosis of chronic villitis can be complicated by villous changes occurring at the periphery of infarcts, or by an unusual villous stromal appearance in some cases of prematurity. Some authors have attempted to define immunohistochemical criteria to facilitate the sensitivity and reproducibility of this diagnosis.59 However, this has not yielded the hoped-for gold standard.60 A conservative route may avoid some of the pitfalls and variability in the diagnosis of chronic villitis. Only the four sections of grossly normal villous parenchyma are searched for chronic villitis, eliminating the question of ischemic change versus villitis at the periphery of infarcts or other lesions. The issue of stromal cellularity versus chronic villitis arises infrequently and may be resolved by lymphocyte-specific immunohistochemistry. Chronic intervillositis may be more readily diagnosed in preterm placentas than chronic villitis. The relative volume and spatial extent of villous involvement is conveyed by grading. Grade 1 is a solitary focus of chronic villitis in any one of four slides, and grade 2 two foci of chronic villitis on one slide. Higher grades are assigned subjectively based on estimated percentage of inflamed villi. Chronic villitis is seen in about 15% of normal term placentas. More than 90% are grade 1 or grade 2. In uncomplicated term deliveries, more severe grades of chronic villitis are uncommon. Correlation between the grade of chronic villitis and the severity of pregnancy compromise may be inconsistent. Russell54 suggested that the degree of growth restriction was proportional to the degree of villitis, but this was not our experience in a community hospital-based population.60
Chronic lymphohistiocytic inflammation of the intervillous space is termed chronic intervillositis.56 Chronic intervillositis is seen in maternal malaria.61 In this country, it is more often seen in cases of maternal immunologic diseases, in which it may represent a maternal response to placental alloantigens or autoantigens common to mother and placenta.56 At term, it is almost always seen with chronic villitis but is more commonly an isolated finding in preterm placentas. This may be due to the immunologic immaturity of preterm villous stromal macrophages, with reduced expression of adhesion molecules62 and other phenotypic markers of monocyte maturation,63 making the development of a focal villous inflammatory infiltrate less likely. Chronic intervillositis is often associated with trophoblastic necrosis. Many of the intervillous cells are reactive immunohistochemically for IgM, IgG, and/or IgA, which may suggest intervillous immune complex formation (and their clearance by intervillous macrophages). Certain antiphospholipid antibodies have been shown to be cross-reactive to trophoblast phospholipid epitopes. In cases of fetal growth restriction with chronic villitis,64 it may be tempting to speculate that chronic villitis represents the effects of alloreactivity of autoantibodies, with trophoblast damage and lymphocyte recruitment.65 It is not surprising, therefore, that chronic intervillositis has been observed in recurrent pregnancy loss.66, 67 Acute intervillositis is seen with maternal sepsis or severe intra-amniotic infection.
Some decidual lymphocytes are normally present in the decidua of the basal plate and the extraplacental membranes. “Chronic choriodeciduitis/amnionitis” implies not only a denser distribution of decidual lymphocytes than “normal” but also the migration of (maternal) decidual lymphocytes into the fetal tissues of the chorion and even the amnion. In uncomplicated term deliveries, we have found decidual plasma cells to be uncommon, which may be reasonable given the potential effects of antibody-forming cells in the basal plate juxtaposed to fetal placental cells. Decidual plasma cells in extraplacental membranes and/or the basal plate are not specific to any one complication of pregnancy, being commonly seen in severe Rh isoimmunization, antiphospholipid antibody-related pregnancy compromise, cytomegalovirus, herpes and syphilis, and “treated” intra-amniotic bacterial infections (especially in the context of attempted tocolysis). Decidual plasma cells also are present with defective trophoblast conversion of uteroplacental arteries and maternal immunologic disorders. In the basal plate, dense chronic decidual inflammation may accompany uteroplacental vascular lesions. Chronic uteroplacental vasculitis is significantly more common in early euploid pregnancy loss than in aneuploid pregnancy loss.68 The same lesion (chronic uteroplacental vasculitis) is among the lesions associated with low-birthweight infants.69 Differences in clinical outcome with identical histopathology may be best explained by different causes of inflammation, different severities of inflammatory stimulus, and host response. For example, a correlation among levels of antiphospholipid antibodies, chronic uteroplacental vasculitis, and poor pregnancy outcome has been suggested.70 This pathway suggests that antiphospholipid antibodies directly induce pregnancy failure by way of induction of deleterious cellular immune responses rather than by initiation of coagulation. Isolated (focal) chronic uteroplacental vasculitis is also the most common uteroplacental vascular lesion at term.71 There may be a greater tendency to develop chronic inflammatory lesions with increasing gestational age, based in part on maturation of the placental macrophage and in part on the cumulative effects of syncytial damage (reflected by increased amounts of perivillous fibrin/fibrinoid) and capillary damage (reflected by a greater incidence of fetomaternal transfusion in the last trimester). Our current studies support the proposition that although no lesion is specific to any particular form of pregnancy compromise, patterns of related lesions are highly distinctive for different clinical outcomes and show strong associations with gestational age at delivery.72, 73
ABNORMAL MATERNAL–PLACENTAL PERFUSION (UTEROPLACENTAL VASCULAR PATHOLOGY)
Physiology of abnormal uteroplacental vascular conversion
An outline of the processes of conversion of the uteroplacental vasculature from a high-resistance/low-capacitance circuit to a high-capacitance system able to carry large volumes of blood to the intervillous space is presented in other chapters in this library. During pregnancy, adaptive changes associated with endovascular trophoblast invasion, which occurs in most spiral arteries, lead to dramatic increases in diameter.74 In complicated pregnancies, this vascular adaptation takes place only in a limited number of placental bed arteries. Failure of uteroplacental vascular conversion is not specific for preeclampsia or even for hypertensive disease in pregnancy. Abnormal uteroplacental vascular conversion and uteroplacental vascular damage also underlie many spontaneous preterm births.69, 75, 76 It is reasonable to speculate that abnormal uteroplacental vascular conversion (and hence uteroplacental perfusion) is the primary process underlying endothelial cell activation in preeclampsia77 or causing fetal nutritional deprivation in fetal growth retardation and some cases of intrauterine fetal demise. A causal pathway relating uteroplacental vascular pathology to preterm labor and premature membrane rupture has been hypothesized.78 It is critical to keep in mind that the pathologic division between preeclamptic and nonhypertensive complications of pregnancy may not always be firm. Likewise, there is a spectrum of uteroplacental vascular changes that extends from the severely compromised growth-restricted preterm infant to the term, low ponderal index infant, to the infant electively delivered due to reduced tolerance of labor. A recent study79 found “pathological changes” in 58% of complicated pregnancies but also in 40% of normal pregnancies. The authors cited sampling error as a possible explanation, because a typical biopsy contains only one or two spiral arteries. This underscores the advantages of generous placental sampling, which provides a broader view of the uteroplacental environment. These authors also suggest that additional factors besides uteroplacental vascular lesions might be required to induce clinical preeclampsia. This is consistent with the variety of pathways to preeclampsia recently proposed by Redman and Sargent.80 These pathways include oxidative placental damage and chronic inflammatory placental lesions, each leading to activation of the maternal endothelium, but also providing for preeclampsia developing in the context of a completely normal placenta when the mother has a chronic endothelial pathology. This last is more common in term preeclampsia, in which chronic uteroplacental vascular lesions can be uncommon.81 No uteroplacental arterial lesion is distinguished among the different clinical presentations associated with uteroplacental vascular pathology.69, 82 No single uteroplacental histologic feature uniquely characterizes preeclampsia as opposed to, for example, fetal growth retardation.82, 83 Spiral and basal arteries of the placental bed may be more tortuous and/or densely distributed in preeclamptic than normal term placental bed arteries.83 Normal placental expansion in the early trimesters stretches the placental bed arteries.84 The increased tortuosity we observed may be due to reduced placental growth preeclampsia, with limited expansion of the uterine cavity leaving “redundant” (and therefore tortuous) spiral vessels. This would lead to a hemodynamically vulnerable architecture at greater risk for shear stress-induced endothelial injury. In the presence of physiologic vasoconstriction and/or myointimal hypertrophy, the poor hemodynamic situation would become worse, in a vicious cycle of vascular injury. Pathologic implantation may also deform the normal complex quadratic distribution of interstitial trophoblast invasion,85 which may parallel endovascular conversion in normal pregnancy.
Another mechanism for vascular injury in failed conversion may be related to the observation that arterial wall disorganization occurs to some degree before there is intravascular or even local trophoblast.86 If these early vascular modifications occur but are not followed by trophoblast invasion and vascular remodeling, the spiral arteries may be unstable and vulnerable to flow-mediated injury. While purely mechanical forces may stress the uteroplacental circulation, molecular signals may also contribute to the genesis or evolution of vascular lesions. Cytotrophoblast cells isolated from the placenta and placental bed in cases of fetal growth retardation have been shown to express significantly higher levels of plasminogen activator inhibitor-1. This may reduce placental and uteroplacental arterial capacity to lyse fibrin and has been proposed as a mechanism for restricting endovascular conversion (by obstruction) and increasing perivillous fibrin deposition.87 Some cytotrophoblasts in preeclampsia have been reported to have increased xanthine oxidase activity and decreased expression of superoxide dismutase that would shift the local balance in favor of increased reactive oxygen species. The associated finding of peroxynitrite deposition suggests local superoxide/nitric oxide interactions that may reduce vascular responsiveness to normal modulators.88 Finally, the contributions of maternal systemic pathologies to uteroplacental vascular disease cannot be ignored. Given the unique circumstances of spiral arterial remodeling, it might be expected that maternal circulating atherogenic factors might preferentially initiate uteroplacental injury by depositing within the fibrinoid, or compromising endothelial regrowth.
Histologic markers of systemic vascular damage such as lipoprotein(a)89 or endothelial activation90 have been widely studied, but primarily in preeclamptic pregnancies. Given the wide range of pathophysiologies that can lead down the final common pathway of preeclampsia, it is not surprising that there is little consensus. Lipoprotein(a) may be a good example of the diverse clinical manifestations of vascular injury and the diverse uses of hemostatic molecules in normal pregnancy. Berg and associates91 reported women with very high serum lipoprotein(a) levels delivering consecutive very-low-birthweight infants with placentas described as “small and ischemic.” Meekens and coworkers92 found lipoprotein(a) deposition in the placental bed spiral arteries to be increased in preeclamptic cases compared with normotensive controls. This deposition appeared to be directly proportional to the severity of histologic spiral arterial injury. Further work included placental basal plate arteries in normal term births, term preeclampsia, preterm preeclampsia, spontaneous preterm birth, and postpartum curettage and peripartum hysterectomy samples (Table 1).93
Table 1. Distribution of lipoprotein(a) immunoreactivity in uteroplacental vessels of the placental bed and basal plate
No. of Lipoprotein(a)-Reactive Uteroplacental Spiral Arteries | p Value | OR (95% CI) | |
| Placental bed biopsy | | ||
| Normotensive | 2/69 (3%) | ||
| Preeclampsia | 71/179 (40%) | <0.0001* | 22 (5–134) |
| Basal plate | |||
| Normal term | 14/35 (40%) | ||
| Term preeclampsia | 20/21 (95%) | 0.0001† | 30 (3.5–670) |
| Preterm preeclampsia | 10/10 (100%) | 0.003† | 150 (NA) |
| Spontaneous prematurity | 49/56 (88%) | <0.0001† | 10.5 (3–34) |
| Remote postpartum implantation site | 65/70 (93%) | 0.0001† | 19.5 (6–72) |
| Massive peripartum hemorrhage | 0/40 (0%) | <0.0001‡ | >1000 (NA) |
*Significance compared with normotensive placental bed biopsies.
†Significance compared with normal term basal plate.
‡Significance compared with involuting implantation sites.
CI, confidence interval; NA, not applicable; OR, odds ratio.
Normal uteroplacental vascular involution after placenta delivery is accompanied by thrombosis, leukocytic infiltration of the vascular wall, and proliferation/migration of endothelia and vascular smooth muscle.94 Similar processes occur in atherosclerosis, where platelets, lymphocytes, and mononuclear leukocytes are recruited to the artery wall and change the vascular microenvironment.95 Andrew and colleagues94 observed that complement and other markers of potential vascular pathology were present in the normal term placental bed arterial wall. They described “invariably” the presence of conglutinated erythrocytes and/or thrombus. In implantation sites obtained remote from delivery, arterial lipoprotein(a) deposition was a constant feature. We saw no thrombi in the two cases with immediate, intractable postpartum hemorrhage, and lipoprotein(a) deposition was entirely absent. Some preparation of the placental bed, including development of a more thrombogenic vascular environment, may be critical for normal placental separation to occur without maternal exsanguination.
Lipoprotein(a) has been recently been described as “one of the few risk factors capable of promoting both early and advanced stages of atherogenesis,”96 and therefore it may be particularly useful in the transition from free flow to no flow after placental delivery. How might the proposed “vascular preparation” develop? The mechanical trauma of labor, compressing and releasing uteroplacental arteries, may initiate vascular damage by shear stress effects as well as basic structural deformation. If lipoprotein(a) deposition occurs within the restricted period from labor to delivery, the smooth muscle cell deposition characteristic of chronic vascular damage might not be expected.97 Our cases of uncomplicated term births delivered after labor uniformly showed dense medial (fibrinoid) deposition. Perhaps the gradual increase in uterine activity before frank labor98 contributes to the vascular changes common to healthy parturition. Precocious or more extensive antepartum development of uteroplacental involution may account for decreased fetal growth velocity in the third trimester. Likewise, the more extensive lipoprotein(a) in the uteroplacental vasculature of term preeclampsia (see Table 1) may result in placental ischemia, as has been proposed as the trigger of the maternal symptoms of preeclampsia.80, 99
Lipoprotein(a) deposition in basal plate arteries was also seen in each of the three types of prematurity studied (preeclampsia, preterm labor, and premature rupture of membranes; see Table 1). In preterm preeclampsia, placental bed arteries with failed trophoblast invasion were uniformly lipoprotein(a) reactive. Lipoprotein(a) deposition may be facilitated by preexisting vascular damage. All of our patients with preterm labor and premature membrane rupture were tocolyzed (prolonging the period of clinical myometrial activity). Therefore, mechanical trauma to the uteroplacental vasculature would be assumed. Despite this, placentas from preterm labor and premature membrane rupture had fewer and less severe uteroplacental lesions and less widespread placental ischemic damage than placentas from preterm preeclampsia.82 Lipoprotein(a) deposition in preterm labor and premature membrane rupture may be the result of chronic low-level uteroplacental vascular pathology, the mechanical trauma of labor, or a combination of the two processes.
A significant role of lipoprotein(a) in processes related to uteroplacental vascular wall damage would explain the recent clinical observation of homocysteinuria in patients with preeclampsia.100 Homocysteine has been studied as a marker of atherosclerotic vascular processes. Harpel and Borth101 have shown that sulfhydryl-containing compounds (including homocysteine) increase the affinity (up to 84-fold) between lipoprotein(a) and fibrin. One group found that the C677T mutation in the methylene tetrahydrofolate reductase gene (which slows homocysteine metabolism) carried a 2.5-fold (confidence interval 1.0–6.0) to 3.29-fold (confidence interval 1.03–10.5) odds of placental vasculopathy.102, 103 When other procoagulant risk factors (such as protein C deficiency or resistance) are screened, presence of the C677T mutation and decreased protein C function bring an additive risk of placental vasculopathy (3.4, confidence interval 1.8–6.42).103 The C677T gene mutation appears to affect the uteroplacental vasculature via elevations of maternal serum homocysteine.104 Plasma levels of homocysteine may be higher in women with preeclampsia,105 possibly reducing the maternal systemic endothelial threshold independent of placental vascular disease, as proposed by Redman and Sargent.80 Uteroplacental vascular disease may be more common in this condition; 31% (26/84) of women with placental infarct or abruption were found to have hyperhomocysteinemia compared with 9% (4/46) of controls.106 Many other heritable risk factors for cardiovascular disease, including factor V Leiden, methylenetetrahydrofolate reductase, and prothrombin gene, and for deficiencies of protein C, protein S, and antithrombin III have also been identified as more prevalent among women with severe preeclampsia. Among women with obstetric vascular complications, a threefold greater prevalence of at least one of these heritable factors or acquired anticardiolipin antibodies has been described, and a more than tenfold increase in “double hits” (presence of two thrombophilic factors).107 The same group studied obstetric vascular complications more broadly, including severe preeclampsia, abruptio placentae, fetal growth restriction, and stillbirth. Fifty-seven patients had a thrombophilic mutation, as compared with 19 women with normal pregnancies (52% and 17%, respectively; p <0.001). Deficiency of protein S, protein C, or antithrombin III or anticardiolipin antibodies was found in 14 additional patients compared with one control (13% and 1%, respectively; p <0.001).108 However, not all reports are so conclusive.109, 110, 111 There are several likely explanations: first, that these factors are not associated with vascular complications in obstetrics, no more than they are in adult vascular disease;112 second, that there is population heterogeneity for the known gene polymorphisms,113 so that prevalence of specific polymorphisms will naturally vary widely, and population-specific polymorphisms may not be identified; and third, that thrombophilia is a multigenic disease.114
It is unlikely that current laboratory testing can label every biologic variable that can predispose to clotting. It is also unlikely that every functional mutation of those genes known to alter hemostasis has been identified in every racial or ethnic group. As of 1997, 650 functional mutations of the cystic fibrosis gene had been identified.115 Only a handful of polymorphisms have been identified in the candidate thrombophilia genes (see above). Finally, the maternal environment likely contributes to vascular fragility, including older maternal age and stress. A “thrombophilia workup” is fast becoming common in the at-risk pregnancy evaluation. In the absence of a positive family history for early-onset cardiovascular disease and placental evidence of uteroplacental vascular pathology, this may not be cost-effective. Given the prevalence of many of these polymorphisms, broad application of thrombophilia screening may result in unnecessary therapy and anticoagulation of asymptomatic carriers whose pregnancy pathology may lie elsewhere. Placental pathology can be a useful screen to select those who might most benefit from a thrombophilia screen. It can also identify patients with vascular pathology that cannot currently be labeled with our laboratory methods who may yet benefit from vascular-directed therapy and surveillance.
Pathology of abnormal uteroplacental vascular conversion
Placental bed biopsy specimens are not easy to obtain, and their interpretation may be even more complicated than placenta histopathologic examination. In the clinical setting, some questions that are critical to the obstetrician are: Is there histologic evidence of uteroplacental vascular pathology? Is the pathology (in the most general of terms) “acute” rather than “chronic”? Is there associated placental damage that may have impaired placental function (and therefore fetal well-being)? These questions can all be answered by careful examination of the delivered placenta, without a placental bed biopsy. The delivered placenta has several advantages over the placental bed biopsy; failing a hysterectomy, biopsy of the placental bed of necessity provides only a small and focal sample of the placental bed, within which there is much regional variation.85 Four or five en face slices of the basal plate (which can all fit into a single cassette) may include the distal (basal plate) segments of several different uteroplacental arteries, located at different sites in the placental bed.116 Variation in uteroplacental vascular anatomy and pathology implies also that one section of the placental villi may not be representative of placental function. Thorough examination of the delivered placenta can provide a better picture of the intrauterine environment of the fetoplacental unit than a placental bed biopsy. Lesions of the end-vascular distribution of the uteroplacental circulation (in the basal plate) may not perfectly mirror myometrial pathology. The ability to routinely identify failure of uteroplacental vascular adaptation, fibrinoid necrosis/atherosis, persistence of endovascular trophoblasts, thrombosis, and chronic vasculitis in the basal plate can clarify the nature and mechanisms involved in pregnancy compromise. For example, in a recent study of women at risk for pregnancy loss and spontaneous preterm birth, both acute ascending infection and decidual vascular lesions were identified. These two patterns were found to segregate with the clinical change of the cervix; those with progressive cervical shortening had significantly more acute inflammation, whereas those without cervical shortening had primarily decidual vascular lesions.117 It can be useful to remember that acute inflammatory and decidual vascular lesions often tend to be mutually exclusive. Arias and colleagues118 proposed two “distinct subgroups among patients with preterm labor and preterm ruptured membranes.” Factor72 and cluster analyses119 have confirmed the tendency for these lesions to segregate in separate subgroups of preterm infants. This distinction can be routinely made by reviewing basal plate uteroplacental vessels whenever a placenta is referred for pathology examination (Fig. 12).
|

In the basal plate or the placental bed, uteroplacental arteries with absent, incomplete, or failed adaptation have variable persistence of vascular muscle and elastic lamina (Fig. 13). This leads to increased uterine vascular resistance, decreased capacitance, and decreased total blood flow to the placenta. Doppler and isotope studies in the human suggest that uteroplacental flow is decreased to 50–70% of normal, which may explain the often associated fetal growth retardation.120, 121 Increased uterine artery resistance parallels histologic evidence of impaired trophoblast migration.122 It is been often held that trophoblast invasion of the uteroplacental vasculature is completed at the myometrial level by the middle of the second trimester. Trophoblast vascular conversion at those deeper levels commonly continues even into the early third trimester. Endovascular trophoblast in the superficial (basal plate) uteroplacental arteries later than the early midtrimester may reflect abnormal (delayed) uteroplacental vascular conversion. Fibrinoid necrosis of the vessel wall with mural foamy cells (“atherosis,” see Fig. 13) is accompanied by dense lipoprotein(a) deposition within the vascular wall, reflecting a vascular pathology similar to atherosclerosis. Other common uteroplacental arterial lesions are thrombosis and chronic vasculitis. For cases of preeclampsia in which uteroplacental vascular pathology is common, Redline and Patterson123 explored the hypothesis that there is a generalized maturation defect in the extravillous trophoblast that leads to increased accumulation of trophoblast in the superficial layers of the implantation site. Increased thickness of basal cytotrophoblast and increased cytotrophoblast proliferation (marked by immunostaining with proliferating cell nuclear antigen) were seen in preeclamptic placentas 24–40 weeks of age compared to term. Whether trophoblast “pile-up” at the placental/decidual interface reflects an intrinsic invasive defect, or whether their migration is inhibited by maternal factors is not clear. This pile-up is not, in our view, specific to preeclampsia. It is, however, yet another marker of abnormal uteroplacental interaction assessable in the delivered placenta.
|
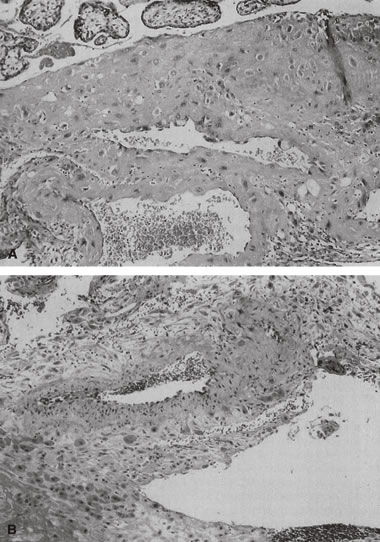
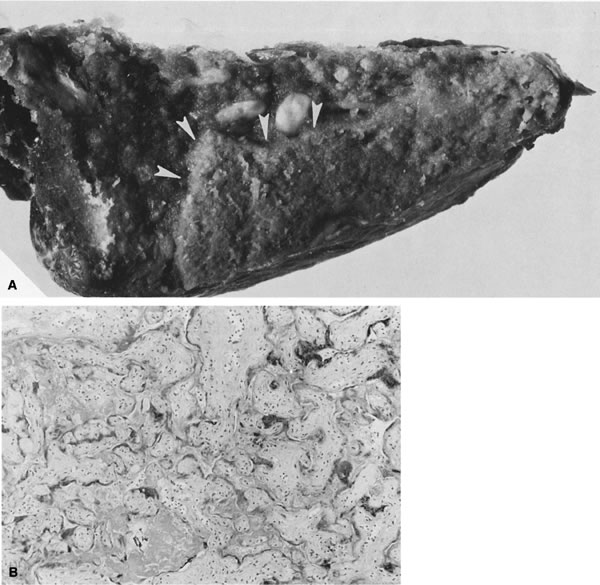
When the uteroplacental vasculature is abnormal, there is a greater chance for “uteroplacental vascular accidents” such as placental infarcts124 or abruption. When uteroplacental arteries are occluded, intervillous flow ceases, the intervillous space collapses, and villi become compressed and undergo ischemic necrosis (an infarct, Fig. 14). Infarcts located in the center of the placenta rather than the perimeter (where there are fewer invasive trophoblasts)85 are considered more significant to the fetus, because the center of the placenta, with optimal trophoblast vascular conversion, should be the most healthy part of the placenta. As many as 10% of completely uncomplicated term births will have one placental infarct, but 90% of these are single, less than 1 cm3 in volume, and located at the placental margin.
|
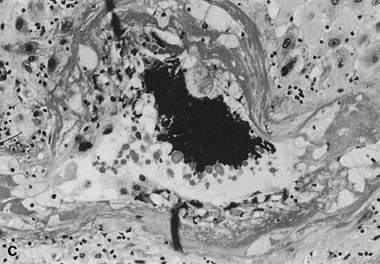
Abruption is a “hemorrhagic infarct.” In abruption, the placenta is forcibly separated from the uterine wall by retroplacental hemorrhage from abnormal uteroplacental vessels.125 Placental compression by a retroplacental hematoma increases fetal blood volume and may be associated with villous stromal hemorrhage (Fig. 15).126 Villous stromal hemorrhage may indicate placental trauma and appears as a bruise. Villous stromal hemorrhage may also be a precursor to fetomaternal transfusion. In our experience, in cases of fetomaternal blood group compatibility (in which preformed maternal antibodies to fetal blood do not exist), an acute abruption that is clinically stabilized may be followed by fetal decompensation as a result of chronic fetomaternal transfusion and severe fetal anemia, which may lead to fetal death. Separation from the uterine lining precludes effective blood flow to the involved placental area, acutely reducing fetoplacental oxygen availability.125 Endothelial damage due to hypoxia is complicated by increased intravascular volume (due to placental compression) to villous stromal hemorrhage.126 This process may also explain the commonly extensive fetal visceral and germinal matrix hemorrhages in abruption.125 Basal intervillous thrombi are primarily maternal blood;127 these lesions may be very mild forms of an abruption-type pathology. Intervillous thrombi off the basal plate may be foci of coagulation initiated by fetomaternal hemorrhage.128 Marginal separation of the placenta may be related to prematurity.129
|

In addition to these large-scale placental lesions, chronically abnormal uteroplacental vascular perfusion may impair the growth and development of the placenta (Fig. 16). Alternatively, it may lead to diffuse villous lesions that cannot be identified grossly. Scarred, shrunken, fibrotic, and hypovascular villi, with reduced placental capillary number and/or caliber, may be caused by capillary destruction by abnormal turbulent uteroplacental flow (Fig. 17).130 Villous capillary damage may lead to fetomaternal hemorrhage. Fetomaternal hemorrhages in the midtrimester (when the barrier between maternal and fetal bloodstreams is still comparatively thick and “sturdy”) are more frequent in hypertensive pregnancies.131 Chronic placental perfusion injury and/or nutritional deprivation may impair terminal villous arborization. The small placenta with uteroplacental vascular lesions may often show histologic increased intervillous volume and decreased villous parenchymal volume, smaller terminal villi, and apparently sparser villous numbers. The placental mass, in these circumstances, may simply fail to develop. The net effect of poor placental growth is a reduced total villous capillary bed. This anatomy would be analogous to an emphysematous lung. Just as significant to the fetus is the effect of reduced placental capillary bed on total peripheral resistance. The smaller placental vascular tree would have increased placental resistance and cause increased cardiac work. Five hundred milliliters per minute of fetal cardiac output is directed to the placenta; the cardiac work effects may be dramatic. An indirect reflection of umbilical-placental resistance is the umbilical systolic/diastolic ratio. This ratio approaches infinity and end-diastolic flow in the umbilical artery may be negative when the placental capillary bed is reduced by more than 50%.132 A reduction in the total fetoplacental capillary bed is paralleled by a reduction in fetoplacental volume. Reduced fetal blood volume can result in a reduced fetal glomerular filtration rate and oligohydramnios.133
|

|
Coagulation
Coagulation is a principal defense mechanism of organisms against invading microorganisms. It plays a central role in both the damage and repair stages of tissue or cell injury or death. Pathologic initiation of coagulation has been used as a marker of clinically significant allograft rejection.134, 135 Coagulopathy has been implicated in the pathogenesis of certain types of obstetric compromise, including antiphospholipid antibody-related fetal death136 and preeclampsia.137 Maternal hypercoagulable states may accompany maternal autoimmune diseases (e.g., systemic lupus erythematosus) or may occur in clinically healthy patients. The increased risk of pregnancy failure in the context of maternal hypercoagulability is independent of maternal clinical disease status.138, 139, 140, 141, 142 The characteristic pathology of pregnancy loss in these conditions (uteroplacental thrombosis and placental infarction)143 is not pathognomonic of any specific laboratory abnormality, although it may be due as much to the inadequacy or incompleteness of our laboratory screening tests for coagulopathy as to the nonspecificity of the pathologic process. For a decade, all patients followed in a university-based rheumatology clinic were prospectively studied. Chronic inflammatory lesions were a significant component of the pathology in mothers with systemic lupus erythematosus, antiphospholipid antibodies, or both. However, the maternal clinical disease and serologic profiles correlated poorly with clinical outcome, with the type of placental pathology (coagulation-related, chronic inflammation, or uteroplacental vascular), and with the site of placental injury. It is not surprising, therefore, that serologic delineation of the syndromes of obstetric compromise is controversial.144, 145 Generally accepted laboratory assays include lupus anticoagulant and anticardiolipin and antiphospholipid antibodies.143, 144, 145, 146 Deficiencies of protein C and S,147, 148 and antithrombin III may cause hypercoagulability during pregnancy, fetal wastage, and similar placental findings. The lack of specificity in the clinical and laboratory assessment of pregnancy outcome and the apparent pathophysiologic mechanism of placental damage probably means that we have not completely delineated all clinical and laboratory markers that are directly causative of obstetric compromise.
The uterine vasculature is particularly susceptible to thrombosis (because its endothelium is normally eroded and the basement membranes and decidual stromal collagen are normally exposed to circulating maternal platelets) for up to at least 24 weeks. Complete conversion includes re-endothelialization of the maternal artery. Failure to accomplish maternal endothelial regrowth over the converted uteroplacental vascular wall of fibrinoid material and embedded trophoblasts is part of the vascular pathology of preeclampsia.149
The mechanisms of action of aspirin may include alterations in both maternal systemic and placental prostacyclin/thromboxane production.150 Not all trials have identified beneficial effects of anticoagulants on pregnancy outcome; this variance may be explained by differences in the therapeutic effects of a uniform dosage within a study population151 or by the reduced efficacy of therapy instituted late in the pathophysiologic process.152, 153 Therapy begun after there is a clinically detectable disorder may not be able to normalize uteroplacental perfusion in an abnormally developed vascular anatomy. If the early development of the fetoplacental unit is compromised, it may be impossible for later therapy to yield normal placental (and fetal) growth and development. The reports of potentially deleterious effects, such as an increased risk of fetal death after placental abruption, are of greater concern.154, 155 One study (in which abruption was self-reported) did not confirm an increased risk.156
A limitation of most clinical studies of anticoagulant therapy in pregnancy is the lack of placental histopathologic examination. In one study, data suggested that clinical outcome was improved after aspirin therapy, although no improvement in placental histopathology was noted.157 If the results of this study are confirmed, therapeutic interventions that alter the maternal course but do not correct the intrauterine pathophysiology may require extremely careful fetal monitoring to avoid fetal compromise. If therapy also improves fetal hemodynamics and hence fetal ability to compensate for uteroplacental pathology,158 then less concern about subclinical fetal compromise is warranted.
In our analyses of pregnancies before and after anticoagulant therapy, we devised a diagnostic schema that provides a semiquantitative score of the severity of the lesion and identifies a target tissue for pathologic coagulation. The potential targets for pathologic coagulation include the (maternal) uteroplacental vasculature, the basal plate (including Nitabuch's fibrin), the intervillous space, the villous (syncytiotrophoblast) surface, and the fetoplacental vasculature. In our experience, single thrombotic lesions in the uteroplacental arteries that occlude less than 50% of the lumen and are not accompanied by villous evidence of abnormal uteroplacental perfusion are not uncommon at term. In fact, these lesions may represent part of the uteroplacental vascular preparation for parturition, which allows for rapid cessation of uteroplacental flow at placental delivery and protects the mother against exsanguination.87 Multifocal thromboses completely occluding the uteroplacental arterial lumina are not consistent with uncomplicated term delivery. It has been our experience that maternal anticoagulant therapy is most effective when the maternal vasculature is the target of pathologic coagulopathy, and that it is specifically not effective when coagulation is initiated on the villous trophoblast surface or within the fetoplacental vasculature.
The laying down of a fibrinoid layer at the maternal/placental interface is a standard process of normal pregnancy. This layer of Nitabuch's fibrin is progressively laid down throughout gestation, leading to, at term, a smooth plane of cleavage for the delivering placenta. Even this “normal” amount of coagulation in the basal plate has been proposed to occur as a result of maternal/placental immunologic interactions. The authors also identified increased basal plate coagulation in cases of pregnancy compromise believed to be of immunologic origin.159 This increase in basal coagulation results in a thick band of basal plate fibrin/fibrinoid, with up to several layers of entrapped and variably well-preserved basal villi, often with focal villitis. We have often seen a subjective increase in basal cytotrophoblasts in such cases, a finding that may be reflected in the observations by Redline and Patterson123 of increased basal cytotrophoblast in preeclampsia. We speculate that although an intrinsic trophoblast defect cannot be ruled out, trophoblast invasion is impaired by the markedly abnormal basal matrix. It is also possible that deposition of Nitabuch's fibrin occurs in response to basal cytotrophoblast. Much further work is required to dissect the causal pathways of defective placentation and pregnancy compromise. A highly unusual placental lesion, maternal floor infarction (Fig. 18), shares the histologic features of uteroplacental and decidual “sparing” and intervillous fibrin/fibrinoid but has a much greater mass, generally involving 80–95% of the villous parenchyma. We have seen potential intermediate lesions in the central basal plate: irregularly increased basal fibrin/fibrinoid and five to 15 layers of entrapped necrotic villi in cases of recurrent pregnancy loss. Perhaps maternal floor infarction, a rare lesion recognized to recur in subsequent pregnancies, is an extreme example of a coagulopathy initiated by pathologic maternal/placental interactions in the basal plate. Milder forms of this lesion (in which five to 20-plus villi are entrapped in encroaching parabasal fibrin/fibrinoid) are not uncommonly found in serial pregnancies from women with recurrent pregnancy loss. Anecdotally, this lesion may not be modified by maternal aspirin/heparin therapy.
|
Coagulation on the placental trophoblast surface distant from the basal plate may be initiated by a variety of stimuli, including circulating endotoxin and trophoblast apoptosis,160 and it may play a critical role in syncytial repair. The common finding of placental trophoblast surface coagulation in association with chronic villitis and/or intervillositis supports a hypothesis that inflammatory processes, including local immune complex formation, may damage syncytiotrophoblast. Massive perivillous fibrin/fibrinoid (Fig. 19) with or without chronic placental inflammation is, in our experience, the most common pathology in patients who fail to respond to anticoagulant therapy for antiphospholipid antibody-related pregnancy loss. Some women have never had uteroplacental thromboses in previous (untreated) pregnancies. Other patients have had prior uteroplacental vascular thromboses; in these patients, anticoagulant therapy may have remedied the maternal coagulopathy but not prevented the evolution of coagulopathy at other sites (e.g., the placental surface).
|
Damage to the fetoplacental vasculature commonly results in the initiation of coagulation. One may attempt, with variable success, to distinguish fetoplacental vascular lesions primarily related to coagulation from lesions in which coagulation is secondarily invoked. Thrombotic lesions occurring in the absence of other placental pathology most often involve the chorionic or large fetal stem vessels but can occur at any level of the villous tree. In preterm infants, acute inflammatory thrombi in the amniotic-surface side of the chorionic vessel may be the most common cause of placental vascular thrombosis. In term infants, the most common etiology may vary by patient population. In some populations with a high prevalence of thrombophilic mutations, fetal thrombophilia may be an important contribution to this serious placental lesion. When these lesions are identified in a compromised fetus, detailed maternal and paternal family histories may be useful to clarify inheritance and better estimate recurrence risk in subsequent pregnancies.161 Bland (noninflammatory) eosinophilic mural thrombi are often calcified and grossly visible on careful inspection of the chorionic surface. Such lesions may result from mechanical trauma to the chorionic plate vessels (e.g., fetal kicking of the chorionic plate). In our experience, these lesions are commonly seen in areas of vasa previa and velamentous vessels, if the vessels running in the membranes and/or near the site of rupture are adequately sampled. In cases of acute ascending infection, chorionic plate inflammation and chorionic vasculitis may be accompanied by chorionic mural thrombi, generally on the superficial side of the vessel (closest to the inflammatory stimuli in the amniotic fluid, and in the area of greatest density of neutrophil infiltration). Similarly, bland thrombi can occur in the fetal stem vessels (Fig. 20), although in these cases it is difficult to posit a role of mechanical trauma. These lesions often accompany other evidence of processes that damage endothelia, such as chronic villitis and so-called hemorrhagic endovasculitis. If thrombi in the fetal stem vessels are associated with inflammatory infiltrates, they are most commonly chronic in nature and are associated with chronic villitis in other sites. Many times, vascular lesions may be seen in cases of stillbirth. When vascular lesions are caused by stillbirth (stillbirth being a “global” process), the vascular lesions are more generalized and may begin in the capillary bed. We also believe that “multiple lumens”, recanalization of thrombi, are a marker of antemortem thrombus.
|

Thrombosis of large fetal stem arteries in the placenta results in downstream vascular obliteration and avascular villi. Thrombosis of large fetal stem veins could also result in placental capillary injury but might involve local capillary rupture due to increased venous pressure from local venous obstruction. Venous thrombi are considered to carry the greater risk for fetal cerebral thromboembolism and cerebral injury.162 Redline and Pappin163 have shown that when avascular villi account for more than 2.5% of the placental parenchyma, there are increased rates of intrauterine growth restriction, abnormal antepartum fetal monitoring test results, oligohydramnios, and maternal coagulopathy. Chronic villitis, membrane hemosiderin, meconium in all three membrane layers, and chorangiosis are also more common. The neonates also had increased immediate morbidity, including thrombotic events. Both mother and neonate must be carefully evaluated when extensive avascular villi are identified.
Abnormal fetoplacental perfusion associated with intraplacental vascular pathology
If the developmental program of the conceptus is normal (normal karyotype) and there is adequate maternal nutrient provision to the conceptus, then an appropriate vasculature to transport the nutrients from the placenta to the fetus proper is still required for successful completion of pregnancy. Placental vasculogenesis and angiogenesis have been reviewed.164 While early villous growth primarily reflects trophoblast differentiation and proliferation, later villous development is marked by extensive terminal villous capillary growth.165
Placental vascular obliterative lesions
Placental vessels can develop normally but then be removed from function by thrombi, which can occur as a primary process or develop as a result of other placental pathophysiology (e.g., chronic villitis). They can also be modified by extrinsic forces (related to maternal perfusion) and remodeled by aberrant flow patterns within the placenta itself. Lesions that either preclude the subsequent normal development of the placental capillary bed or remove parts of it from function would be expected to affect umbilical artery Doppler velocimetry. Indeed, altered placental morphology is observed in patients with abnormal umbilical artery velocity waveforms.166, 167, 168, 169, 170, 171, 172 A reduced number of small muscular arteries in the fetal stem villi165, 166, 167, 168, 169 and more extensive ischemic pathology171 have been reported in patients with abnormal umbilical artery Doppler velocimetry compared with normal controls. These two lesions may be causally related; reduced uteroplacental perfusion can result in uteroplacental ischemia and chronic uteroplacental vasoconstriction.172 However, at least one report170 identified severe chronic villitis associated with abnormal umbilical artery Doppler, suggesting that the pathophysiology of abnormal umbilical artery Doppler may be more complex. We studied 52 consecutive, nonanomalous singletons delivered between January 1989 and June 1995. These infants were in less than the 10th centile for birthweight (fetal growth retardation) and were admitted to the neonatal intensive care unit with umbilical artery Doppler velocimetry obtained within 3 days of delivery.173 We expected that intraplacental vaso-occlusive processes would show a continuum from normal to a reversal of end-diastolic flow. Instead, we identified two major subsets of fetal growth retardation by characterizing them according to the level of intraplacental vaso-occlusive lesions. Lesions considered to reflect intraplacental vascular pathology included the following:
- Mural or occlusive fibrin thrombi in chorionic and fetal stem vessels
- Avascular terminal villi, diagnosed when nutrient villi lacked capillaries and had a dense paucicellular eosinophilic stroma (Fig. 21)
- Hemorrhagic endovasculitis
- Reduction or obliteration of chorionic or fetal stem vessels by mural hyperplasia, identified by a decreased or absent lumen area and increased wall thickness for the level of the fetal vessel (chorionic, large fetal stem artery, small fetal stem artery)
- Mural disorganization, diagnosed when the normal concentric organization of the arterial wall was interrupted over at least 50% of its circumference
- Abnormally thin-walled arteries, diagnosed when all vessels in a large or small fetal stem were histologically similar, implying that the obligatory artery in the fetal stem had vein-like characteristics
|
Baseline levels of placental lesions were defined as the mean number of lesions in fetal growth retardation placentas with normal umbilical artery Doppler waveforms, and excess levels were defined as numbers of vascular lesions that were greater than baseline. The first growth retardation subset defined consisted of those with excess intraplacental lesions. In these cases, there were more uteroplacental vascular lesions, more excess lesion scores, and lighter placentas, and these features were related to worsening umbilical artery Doppler status, consistent with our hypothesis of a continuum of pathologic anatomy from normal umbilical artery Doppler to reversal of end-diastolic flow. These findings support the observations of Arabin and associates169 and Laurini and coworkers,170 who related histologic evidence of uteroplacental ischemia to abnormal umbilical artery Doppler velocimetry. Perfusion of the placenta at abnormally low oxygen tension is associated with increased basal perfusion pressure, consistent with placental vasoconstriction.174 Chronic vasoconstriction (and increased intraluminal pressure) could lead to vascular obliteration by way of progressive mural hyperplasia. However, increased intraluminal pressure could predispose to endothelial damage and luminal obliteration by way of hemorrhagic endovasculitis lesions. We observed histologic changes that could support both of these pathophysiologic mechanisms. We also observed that placentas with intraplacental vaso-occlusive lesions were lighter, implying a diffuse reduction in placental growth as a whole. Others have observed that there is a global reduction or underdevelopment of villous arborization, rather than a selective destruction of one vessel type.175, 176, 177
Macara and colleagues178 showed that terminal villi in cases of fetal growth retardation with absent end-diastolic flow have increased syncytial nuclei, reduced cytotrophoblast nuclei, and thickened basal lamina, with increased stromal collagen and extracellular matrix materials. Their findings suggest that prematurely aged syncytiotrophoblast (with increased nuclei arranged in knots) is less rapidly replenished by cytotrophoblast (reduced cytotrophoblast cell number and proliferation index), which led them to conclude that there was no evidence of villous hypoxia per se but rather a capillary congestion that supports the hypothesis of a primary vasomotor pathology in absent end-diastolic flow. The timely observations of Burton and Jauniaux179 led them to conclude that umbilical vascular resistance may be heavily influenced by the number of parallel circulatory units offered by the placental lobules. In a study of the placental villous response to hypoxia caused by high altitude, maternal iron-deficiency anemia, and preeclampsia, they observed a trend for increased capillary volume, apparently due to capillary dilatation with accompanying thinning of the vasculosyncytial membrane barrier.180
The second group of growth retardation was defined as those cases with baseline intraplacental vaso-occlusive lesions. Among these, an increased chronic inflammation score was related to worsening umbilical artery Doppler status, despite no recognizable increase in intraplacental vaso-occlusive lesions. Chronic inflammation-associated abnormal umbilical artery Doppler waveforms may represent the effects of abnormal vascular function rather than fixed anatomic lesions. Cytokines and prostanoids (known to affect placental vasomotor tone)181 may be generated in the process of chronic inflammation and cause abnormal placental resistance in the absence of anatomic lesions. This distinction could be clinically relevant because malfunction of an anatomically normal placenta may be amenable to in utero therapy (e.g., transplacental administration of anti-inflammatory agents).
It may be too optimistic to expect the range of umbilical artery Doppler from normal to reversal of end-diastolic flow to show a pathologic continuum, but it is reasonable to expect absent and reversed end-diastolic flow to have a close resemblance. Although our findings may show the effects of small sample size, reversed end-diastolic flow cases were striking for the relative paucity of vaso-occlusive lesions and for a significant increase in structures best described as vein-like fetal stem arteries. One explanation may be that some cases of reversed end-diastolic flow represent unique pathophysiologic processes. Alternatively, a pattern of degenerative and/or dysplastic aortic architecture that effectively reverses the normal differentiation pattern of aortic wall development has been reported in cases of twin-reversed arterial perfusion.182 This maldevelopment has been suggested to reflect the effects of abnormal levels and gradients of intraluminal pressure. Whether the embryonal vessels forming within the primitive chorionic membrane of the placenta will become arteries or veins is believed to be “determined by the direction of flow and its pressure”.183 Thus, our observations suggest that in fetal growth retardation with reversed end-diastolic flow, the placental arterial wall structure may be modified (and lesions obscured) by the reversed blood flow. The placental vasculature is a dynamic structure, the development and expansion of which continue throughout most of gestation. The potential for mechanical factors (e.g., intraluminal pressure and flow direction) to remodel and in effect deform the evolving placental vasculature cannot be underestimated. Karimu and Burton184, 185 have confirmed the mechanical effect of intraplacental perfusion pressure on villous capillary growth. They noted that more proliferating endothelial nuclei were found at 100 mmHg than at 40 mmHg. This suggests that mechanical factors may play a role in villous angiogenesis and the formation of terminal villi. They have also shown similar effects of maternal perfusion on the placental capillary bed. The study showed that placental capillaries are elastic and deformable, and also that if a sufficiently high pressure is generated in the intervillous space, they may be compressed. They suggested that this may also provide an explanation for the observed increase in umbilical vascular resistance shown by Doppler studies in pregnant women scanned in the supine position.
Vascular proliferative lesions
Abnormally proliferative placental vascular development is commonly related to maternal diseases involving overgrowth of the placenta as a whole (Fig. 22). Cardinal among these conditions is maternal diabetes mellitus. In maternal diabetes mellitus, abnormal growth factor expression and effect may be associated with overgrowth of the placental vessels, as well as with a wide range of direct fetal effects, including macrosomia, polycythemia, and late fetal death.186 Vascular proliferation can be seen in other conditions, including fetal growth retardation187 and twin gestations. In the latter case, two fetuses are competing for a single volume of uterine blood flow. In maternal diabetes mellitus, fetal growth retardation, and multiple gestation, capillary proliferation may be an endothelial response to decreased oxygen availability. Capillary proliferation may increase placental microvascular resistance and the diffusion distance of nutrient across the placental membrane, and it may increase fetoplacental intravascular volume and fetal cardiac work. We have repeatedly observed in our clinical practice that umbilical systolic/diastolic ratios generally remain within normal limits even when there is a massive increase in villous capillary number, as long as other placental lesions that obliterate the placental vasculature are not present. Chorioangioma, a focal placental vascular tumor or malformation, occurs singly or multifocally and may function as an arteriovenous malformation or vascular shunt (Fig. 23). This lesion may present with stillbirth, hydrops fetalis due to congestive heart failure, or unexplained fetal tachycardia.
|
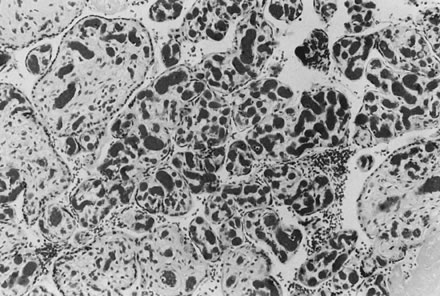
|
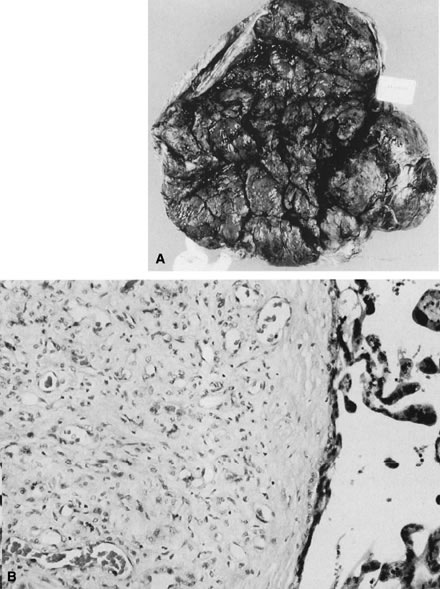
Pattern recognition in placental histopathology: clinical applications in prematurity
A comprehensive prospective data collection on all pregnancies delivered at a university health center formed the basis of a data set of extreme prematurity (deliveries at less than 32 weeks' gestation). As extensively reported elsewhere,14, 73, 79 the study population excluded clinical factors that could be considered predisposing to preterm birth (e.g., multiple gestation, fetal congenital anomaly, maternal diabetes mellitus, and isolated chronic hypertension), conditions resulting in elective preterm birth (e.g., placenta previa or isolated fetal growth restriction with fetal distress), and cases in which there was a discrepancy between the obstetric and neonatal assessments of gestational age. For 432 pregnancies, preterm birth could be attributed to one of three principal indications for delivery: premature membrane rupture, premature labor with intact labor, or preeclampsia. Placental histopathologic features were scored semiquantitatively in five primary pathophysiology categories: acute inflammation, chronic inflammation, uteroplacental vascular pathology and related villous lesions, intraplacental vascular pathology, and coagulation-related pathology. To this data set we applied factor analysis (a statistical technique that extracts complex patterns of correlations within a data set and generates sets of equations that identify interrelations among lesions) and weighted the contribution of the particular lesion to the overall pattern. Essentially, a factor represents a group of variables that are correlated with each other but are relatively uncorrelated with other variables. Our goal in using factor analysis was to reduce the complexity of analyses required in univariate studies of large numbers of specific lesions. We accomplished this by analyzing instead a smaller number of factors and weighting the different lesions within them.
The results of factor analysis of placental histopathologic lesions in preterm preeclampsia have been published.72 Factor analysis generated 15 lesion categories in preterm labor and 15 in premature rupture of membranes that combined different lesions with different strengths of associations (coefficients) among lesions (Table 2 and Table 3). Our goal was to identify patterns that combined several lesions into a single numeric score that would better relate to outcome than any one lesion in isolation. To test this, we examined the relation between the factors and gestational age at delivery. Histopathologic factors generated regression equations highly predictive of gestational age at delivery in both premature membrane rupture (R2 = 0.18) and preterm labor (R2 = 0.28). At least 66% of cases had high scores for more than one factor. We needed to determine whether the placental factors showed additive or synergistic effects on gestational age. For each case, the scores for the categories related to gestational age were summed and plotted against gestational age (Fig. 24). These data suggest that preterm labor or premature rupture of membranes is infrequently due to a single problem; it more often is the result of the application of different “straws” that culminate in “breaking the camel's back” (premature membrane rupture or preterm labor).
|
| Factor 1 | Factor 14 | Factor 8 | Factor 6 | Factor 2 | Factor 4 | Factor 5 | Factor 3 | Factor 7 | Factor 11 | Factor 9 | Factor 10 | Factor 12 | Factor 13 | Factor 15 | * |
| Acute inflammation |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| Acute amnion inflammation | 0.72 |
|
|
|
|
|
|
|
|
|
|
|
|
|
| 0.52 |
| Acute choriodecidual inflammation | 0.86 |
|
|
|
|
|
|
|
|
|
|
|
|
|
| 0.74 |
| Marginating pattern of acute inflammation | 0.71 |
|
|
|
|
|
|
|
|
|
|
|
|
|
| 0.50 |
| Acute chorionitis | 0.87 |
|
|
|
|
|
|
|
|
|
|
|
|
|
| 0.76 |
| Acute chorionic vasculitis | 0.72 | 0.27 |
|
|
|
|
|
|
|
|
|
|
|
|
| 0.59 |
| Acute umbilical vasculitis | 0.72 |
|
|
|
|
|
|
|
|
|
|
|
|
|
| 0.52 |
| Eosinophilia in fetal inflammatory infiltrates | 0.53 | 0.53 |
|
|
|
|
|
|
|
|
|
|
|
|
| 0.56 |
| Uteroplacental vascular pathology | ||||||||||||||||
| Uteroplacental vessel fibrinoid necrosis/atherosis |
|
| 0.49 |
|
|
|
|
|
|
|
|
|
|
|
| 0.24 |
| Uteroplacental vessel—absence of physiologic conversion |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| 0.00 |
| Uteroplacental vessel—persistence of endovascular trophoblast |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| 0.00 |
| Decidual hemosiderin |
|
|
| 0.23 |
|
|
|
|
|
|
|
|
|
|
| 0.05 |
| Placental lesions related to uteroplacental vascular pathology | ||||||||||||||||
| Histology related to abruption |
|
|
| 0.6 |
|
|
|
|
|
|
|
| 0.21 |
|
| 0.40 |
| Villous infarct |
|
| 0.48 | 0.23 |
|
|
|
|
|
| 0.26 | 0.28 |
|
|
| 0.43 |
| Syncytiotrophoblast knotting |
|
|
|
| 0.46 |
|
|
|
|
|
|
|
| 0.27 |
| 0.28 |
| Cytotrophoblast (“X-cell”) proliferation |
|
|
|
|
| 0.36 | 0.37 |
| 0.28 | 0.32 |
|
|
|
|
| 0.45 |
| Villous fibrosis |
|
|
|
| 0.69 |
| 0.21 |
|
|
|
|
|
|
|
| 0.52 |
| Villous hypovascularity |
|
|
|
| 0.77 |
|
|
|
|
|
|
|
|
|
| 0.59 |
| Circulating nucleated erythrocytes |
|
|
| 0.28 |
|
|
|
|
|
|
|
|
|
| 0.21 | 0.12 |
| Chronic inflammation | ||||||||||||||||
| Chronic uteroplacental vasculitis |
|
|
|
|
|
|
| 0.36 |
|
|
|
|
|
|
| 0.13 |
| Decidual eosinophilia |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| 0.00 |
| Basal plate plasma cell infiltrates |
|
|
|
|
|
|
| 0.41 |
|
|
|
|
|
|
| 0.17 |
| Chronic basal plate inflammation |
|
|
|
|
|
|
| 0.78 |
|
|
|
|
|
|
| 0.61 |
| Villitis of anchoring villi |
|
|
|
|
|
|
| 0.62 |
|
|
|
|
|
|
| 0.38 |
| Chronic intervillositis |
|
| 0.61 |
|
|
|
|
|
|
|
|
|
|
|
| 0.37 |
| Chronic villitis |
|
| 0.21 |
|
|
|
|
| 0.47 |
|
|
|
|
|
| 0.27 |
| Chronic chorionitis/amnionitis |
|
|
|
|
|
|
|
|
| 0.63 |
|
|
|
|
| 0.40 |
| Coagulation-related lesions | ||||||||||||||||
| Uteroplacental vessel thrombosis |
|
|
|
|
|
|
|
|
|
|
| 0.79 |
|
|
| 0.62 |
| Intervillous thrombus |
|
|
|
|
|
|
|
|
|
| 0.47 |
|
|
|
| 0.22 |
| Perivillous fibrin deposition |
|
|
|
|
|
| 0.94 |
|
|
|
|
|
|
|
| 0.88 |
| Intervillous thrombus with peripheral infarct |
|
|
|
|
|
|
|
|
|
| 0.9 | 0.2 |
|
|
| 0.85 |
| Villous stromal mineralization |
|
|
|
|
| 0.72 |
|
|
|
|
|
| 0.33 |
|
| 0.63 |
| Chorionic vessel thrombus |
|
|
|
|
| 0.25 |
|
|
|
|
|
| 0.26 |
| 0.47 | 0.35 |
| Fetal stem vessel thrombus |
|
|
|
|
|
|
|
|
|
|
|
| 0.61 |
|
| 0.37 |
| Hemorrhagic endovasculitis |
|
|
|
|
| 0.69 |
|
|
|
|
|
|
|
|
| 0.48 |
| Avascular terminal villi |
|
|
|
|
|
|
|
| 0.8 |
|
|
|
|
|
| 0.64 |
| Miscellaneous (unclassified) lesions | ||||||||||||||||
| Basal plate myometrial fibers |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| 0.00 |
| Laminar chorionic necrosis |
|
|
|
|
|
|
| 0.22 |
|
|
|
|
|
|
| 0.05 |
| Placental “growth zones” evident |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| Villous hypervascularity |
|
|
|
|
|
|
|
| 0.21 | 0.23 |
|
|
|
|
| 0.10 |
| Villous edema |
|
|
|
|
|
|
|
|
|
|
|
|
|
| 0.27 | 0.07 |
| Villous stromal hemorrhage |
|
|
| 0.67 |
|
|
|
|
|
|
|
|
|
|
| 0.45 |
| Villous microcalcification |
|
|
|
|
|
|
|
|
|
|
|
|
| 0.66 |
| 0.4 |
*Variable communality.
|
| Factor 1 | Factor 2 | Factor 3 | Factor 4 | Factor 5 | Factor 6 | Factor 7 | Factor 8 | Factor 9 | Factor 10 | Factor 11 | Factor 12 | Factor 13 | Factor 14 | Factor 15 | * |
| Acute inflammation | ||||||||||||||||
| Acute amnion inflammation | 0.85 |
|
|
|
|
|
|
|
|
|
|
|
|
|
| 0.72 |
| Acute choriodecidual inflammation | 0.83 |
|
|
|
|
|
|
|
|
|
|
|
|
|
| 0.69 |
| Marginating pattern of acute inflammation | 0.63 |
|
|
|
|
|
|
|
|
|
|
|
|
|
| 0.40 |
| Acute chorionitis | 0.82 |
|
|
|
|
|
|
|
|
|
|
|
|
|
| 0.67 |
| Acute chorionic vasculitis | 0.85 |
|
|
|
|
|
|
|
|
|
|
|
|
|
| 0.72 |
| Acute umbilical vasculitis | 0.73 |
|
|
|
|
|
|
|
|
|
|
|
|
|
| 0.53 |
| Eosinophilia in fetal inflammatory infiltrates | 0.6 | 0.32 |
|
|
|
|
|
|
|
|
|
|
|
|
| 0.46 |
| Uteroplacental vascular pathology | ||||||||||||||||
| Uteroplacental vessel fibrinoid necrosis/atherosis |
| 0.71 |
|
|
|
|
|
|
|
|
|
|
|
|
| 0.50 |
| Uteroplacental vessel—absence of physiologic conversion |
|
| 0.69 |
|
|
|
|
|
|
|
|
|
|
|
| 0.48 |
| Uteroplacental vessel—persistence of endovascular trophoblast |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| 0.00 |
| Decidual hemosiderin |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| 0.00 |
| Placental lesions related to uteroplacental vascular pathology | ||||||||||||||||
| Histology related to abruption |
|
|
|
|
|
| 0.63 | 0.33 |
|
|
|
|
|
|
| 0.51 |
| Villous infarct |
|
|
|
| 0.32 |
|
| 0.67 |
|
|
|
|
|
|
| 0.55 |
| Syncytiotrophoblast knotting |
|
|
| 0.68 |
|
|
|
|
|
|
|
|
|
|
| 0.46 |
| Cytotrophoblast (“X-cell”) proliferation |
|
|
| 0.2 |
| 0.37 | 0.21 |
| 0.44 |
|
|
|
|
|
| 0.41 |
| Villous fibrosis |
|
|
| 0.63 |
|
|
|
|
|
|
|
|
|
|
| 0.40 |
| Villous hypovascularity |
|
|
| 0.72 |
|
|
|
|
|
|
|
|
|
|
| 0.52 |
| Circulating nucleated erythrocytes |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| 0.00 |
| Chronic inflammation | ||||||||||||||||
| Chronic uteroplacental vasculitis |
|
| 0.38 |
|
|
|
|
|
| 0.21 |
|
|
|
|
| 0.19 |
| Decidual eosinophilia |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| 0.00 |
| Basal plate plasma cell infiltrates |
|
|
|
|
|
|
|
|
| 0.8 |
|
|
|
|
| 0.64 |
| Chronic basal plate inflammation |
|
| 0.23 |
|
|
|
|
|
| 0.58 | 0.58 |
|
|
|
| 0.73 |
| Villitis of anchoring villi |
|
|
|
|
|
|
|
|
|
| 0.71 | 0.71 |
|
|
| 1.01 |
| Chronic intervillositis |
|
|
|
|
|
|
|
|
|
|
|
| 0.65 |
|
| 0.42 |
| Chronic villitis |
|
|
|
|
|
|
|
|
|
|
|
| 0.59 |
|
| 0.35 |
| Chronic chorionitis/amnionitis |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| 0.00 |
| Coagulation-related lesions | ||||||||||||||||
| Uteroplacental vessel thrombosis |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| 0.00 |
| Intervillous thrombus |
|
|
|
| 0.35 |
|
|
|
|
|
|
|
|
|
| 0.12 |
| Perivillous fibrin deposition |
|
|
|
|
| 0.89 |
|
|
|
|
|
|
|
|
| 0.79 |
| Intervillous thrombus with peripheral infarct |
|
|
|
| 0.91 |
|
|
|
|
|
|
|
|
|
| 0.83 |
| Villous stromal mineralization |
|
|
|
|
|
|
| 0.3 |
|
|
|
|
| 0.55 |
| 0.39 |
| Chorionic vessel thrombus | 0.4 |
|
|
|
|
|
| 0.21 |
|
|
|
|
|
|
| 0.20 |
| Fetal stem vessel thrombus |
|
|
|
|
|
|
| 0.79 |
|
|
|
|
|
|
| 0.62 |
| Hemorrhagic endovasculitis |
| 0.61 |
|
|
|
|
|
|
|
|
|
|
| 0.28 |
| 0.45 |
| Avascular terminal villi |
|
|
|
|
|
|
|
|
|
|
|
|
| 0.65 |
| 0.42 |
| Miscellaneous (unclassified) lesions | ||||||||||||||||
| Basal plate myometrial fibers |
|
|
|
|
|
| 0.22 |
|
|
|
|
|
|
|
| 0.05 |
| Laminar chorionic necrosis |
|
|
|
|
|
|
|
| 0.3 |
|
|
|
|
|
| 0.09 |
| Placental “growth zones” evident |
|
|
|
|
|
|
|
|
|
|
|
|
|
| 0.61 | 0.37 |
| Villous hypervascularity |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| 0.00 |
| Villous edema |
|
|
|
|
| 0.23 |
|
|
|
|
| 0.43 |
|
|
| 0.24 |
| Villous stromal hemorrhage |
|
|
|
|
|
| 0.64 |
|
|
|
|
|
|
|
| 0.41 |
| Villous microcalcification | 0.69 |
|
| 0.3 |
|
|
|
| 0.34 |
|
|
|
| 0.26 |
| 0.75 |
*Variable communality.
|
The implications of this thesis for current investigations of methods to reduce prematurity (e.g., by antibiotic therapy) are significant. Our analysis indicates that a decrease in amnion inflammation from grade 4 to grade 3 is associated with 1 more day of gestation, whereas a similar unit increase in severity of uteroplacental vascular lesions is associated with 4 fewer days of gestation. The underlying pathophysiology of prematurity in any population and the interplay of the various factors that predispose to prematurity must be carefully taken into consideration in the interpretation of analyses.
The factor categories for premature membrane rupture and preterm labor appear to uniquely characterize these two processes. However, if this is true, the factors and their associations with clinical features should also be unique in the two different outcomes. For both premature membrane rupture and preterm labor, a factor category was generated that primarily represented the effects of histologic markers of acute ascending infection (factor 1; see Table 2 and Table 3), with very similar coefficients. However, when the premature membrane rupture factor of acute inflammation was used to “predict” gestational age in the preterm labor group, it was only 26% as strong a predictor of gestational age as the similar equation generated in the preterm labor data set itself. Although the factors appear to be quite similar, they are in fact quite different and have different associations with such basic clinical concerns as gestational length.
We have applied factor analysis to a series of cases involving infants delivered to women with antiphospholipid antibodies, systemic lupus erythematosus, or both.57 We have also identified patterns of lesions that may contribute to an understanding of the mechanisms of placental damage in these conditions and the differences in patient responses to therapeutic interventions.
In summary, we use a diagnostic approach to placental pathology that is directed toward identifying the underlying pathophysiologic processes (acute inflammation, chronic inflammation, uteroplacental vascular pathology and related villous lesions, intraplacental vascular pathology, and coagulation-related pathology) and the target or targets of tissue injury. The processes and targets revealed can then be related to the clinically manifest pathophysiology of mother and fetus/neonate. This provides, in our opinion, the best mechanism for clinically relevant clinicopathologic correlation. This can also be used in the generation and testing of new hypotheses regarding relations among placental anatomic changes, placental functional disturbances, and maternal and fetal/neonatal outcome.
THE FUTURE OF OBSTETRIC PATHOLOGY
There is so much that is unique to pregnancy—the maternal exposure in such an intimate fashion to paternal antigens, the vascular modeling, the hormonal profile, and the mother's systemic adaptations to the growing conceptus—that it can be overlooked that pregnancy is part of a woman's medical history. During the past decade, evidence from a wide range of studies (clinical, epidemiologic, and animal models) points to the role of the fetal environment in the etiology of adult diseases. Disorders proposed to have their origins in fetal life include hypertension and atherosclerosis,188,189,190 obesity and diabetes,191,192 cancer risk193 (including breast cancer194,195), and neuropsychiatric disorders such as autism196 and schizophrenia.197
Paternal family histories of cardiovascular disease or preeclampsia may not be commonly elicited. The genetic contribution of the father to the trophoblast cell, the lining of the intervillous blood space, and a primary remodeler of the maternal vascular circuit, is thus overlooked. Both epidemiologic198 and placental histologic161 data support that the fetal genotype (inherited from both parents) influences pregnancy outcome and placental growth and development. Ultimately, this effect may echo in the wide range of adult disease risks listed above.
Follow-up studies suggest that women with preeclampsia or who have low-birthweight infants have an increased rate of endothelial dysfunction,199 hypertension, and cardiovascular disease later in life.200 These women also have more commonly family histories of significant cardiovascular disease.199 A woman born from a preeclamptic pregnancy may have a reduced risk of breast cancer in adulthood.201 Certain types of complicated pregnancies may therefore serve as “triage points” to identify women who may benefit from earlier screening and/or treatment for subclinical cardiovascular disease or to modify breast cancer risk assessment.
Birthweight, high or low, is the variable that has been found to influence the disease risks listed above. Not all cases of low birthweight or macrosomia are etiologically identical. It is reasonable that specific risks may be associated with particular etiologic types of low birthweight and macrosomia. Ongoing studies are working to pinpoint the specific mechanisms by which variation in birthweight translates into disease predisposition. The placenta is proving an important tool for the identification and classification of major intrauterine pathophysiologies. Once the causal pathways can be more clearly discerned, study design can be improved to test them more critically. The adult diseases that appear to be influenced by the fetal environment carry enormous public health costs and lost quality of life. Confirmation of the apparent importance of the intrauterine environment to adult disease risk, and clarification of how this relationship is mediated, will be an important step toward improving outcomes of these significant diseases.
REFERENCES
Curzik D, Drazanic A, Hrgovic Z: Nonspecific aerobic vaginitis and pregnancy. Fetal Diagn Ther 16: 187, 2001 |
|
Romero R, Salafia CM, Athanassiadis AP et al: The relationship between acute inflammatory lesions of the preterm placenta and amniotic fluid microbiology. Am J Obstet Gynecol 166: 1382, 1992 |
|
Lyon A: Chronic lung disease of prematurity. The role of intra-uterine infection. Eur J Pediatr 159: 798, 2000 |
|
Berger R, Garnier Y: Perinatal brain injury. J Perinat Med 28: 261, 2000 |
|
Leviton A, Paneth N, Reuss ML et al: Maternal infection, fetal inflammatory response, and brain damage in very low birth weight infants. Developmental Epidemiology Network Investigators. Pediatr Res 46: 566, 1999 |
|
Toti P, De Felice C, Palmeri ML et al: Inflammatory pathogenesis of cortical polymicrogyria: An autopsy study. Pediatr Res 44: 291, 1998 |
|
Yoon BH, Kim CJ, Romero R et al: Experimentally induced intrauterine infection causes fetal brain white matter lesions in rabbits. Am J Obstet Gynecol 177: 797, 1997 |
|
Ghidini A, Salafia CM, Minior VK: Prevalence of intrauterine infection and duration of the latency period in preterm rupture of membranes (abstr 602). Am J Obstet Gynecol 174: 474, 1996 |
|
Romero R, Sirtori M, Oyarzun E et al: Infection and labor: Prevalence, microbiology and clinical significance of intraamniotic infection in women with preterm labor and intact membranes. Am J Obstet Gynecol 16: 817, 1989 |
|
Guzick DS, Winn K: The association of chorioamnionitis with preterm delivery. Obstet Gynecol 65: 11, 1985 |
|
Salafia CM, Mangam HE, Weigl CA et al: Abnormal fetal heart rate patterns and placental inflammation. Am J Obstet Gynecol 160: 140, 1989 |
|
Salafia CM, Silberman L: Placental pathology and abnormal fetal heart rate patterns in gestational diabetes. Pediatr Pathol 9: 513, 1989 |
|
Fleming AD, Salafia CM, Vintzileos AM et al: The relationships amongst umbilical arterial velocimetry, fetal biophysical profile and placental inflammation in preterm premature rupture of the membranes. Am J Obstet Gynecol 164: 38, 1991 |
|
Salafia CM, Ghidini A, Sherer DM, Pezzullo JC: Abnormalities of the fetal heart rate in preterm deliveries are associated with acute intra-amniotic infection. J Soc Gynecol Invest 5: 188, 1998 |
|
Altshuler G: Fusobacteria, an important cause of chorioamnionitis. Arch Pathol Lab Med 109: 739, 1985 |
|
Novak RW, Platt MS: Significance of placental findings in early onset group B streptococcal neonatal sepsis. Clin Pediatr 24: 256, 1985 |
|
Ariel I, Singer DB: Streptococcus viridans infections in mid-gestation. Pediatr Pathol 11: 75, 1991 |
|
Benirschke K, Raphael SI: Candida albicans infection of the amniotic sac. Am J Obstet Gynecol 7: 200, 1958 |
|
Sherman DJ, Tovbin J, Lazarovich T et al: Chorioamnionitis caused by gram-negative bacteria as an etiologic factor in preterm birth. Eur J Clin Microbiol Infect Dis 16: 417, 1997 |
|
Salafia CM, Ghidini A, Sherer DM et al: Fetal, but not maternal, serum cytokine levels correlate with histologic acute placental inflammation. Am J Perinatol 14: 419, 1997 |
|
Burgio GR, Ugazio AG (eds): Immunology of the Neonate. Heidelberg, Springer-Verlag, 1987 |
|
Naeye RL, Maisels J, Lorenz RP et al: The clinical significance of placental villous edema. Pediatrics 71: 588, 1983 |
|
Salafia CM, Minior VK, Rosenkrantz TS et al: Maternal, placental and neonatal associations with early germinal matrix/ intraventricular hemorrhage in infants born at <32 weeks gestation. Am J Perinatol 12: 427, 1995 |
|
Saftlas AF, Olson DR, Atrash HK et al: National trends in the incidence of abruptio placentae, 1979-1987. Obstet Gynecol 78: 1081, 1991 |
|
Ando K, Kanayama N: The study of mechanism of abruptio placentae caused by chorioamnionitis. Nippon Sanka Fujinka Gakkai Zasshi 45: 1035, 1993 |
|
Major CA, de Veciana M, Lewis DF et al: Preterm premature rupture of membranes and abruptio placentae: Is there an association between these pregnancy complications? Am J Obstet Gynecol 172: 672, 1995 |
|
Tolockiene E, Morsing E, Holst E et al: Intrauterine infection may be a major cause of stillbirth in Sweden. Acta Obstet Gynecol Scand 80: 511, 2001 |
|
Staples LD, Heap RB, Wooding FB, King GJ: Migration of leucocytes into the uterus after acute removal of ovarian progesterone during early pregnancy in the sheep. Placenta 4: 339, 1983 |
|
Fox H (ed): Pathology of the Placenta. London, WB Saunders, 1978 |
|
Nelson DM, Crouch EC, Curran EM et al: Trophoblast interaction with fibrin matrix. Am J Pathol 136: 855, 1990 |
|
Frank HG, Malezadeh F, Kertschanska S et al: Immunohistochemistry of two different types of placental fibrinoid. Acta Anat 150: 55, 1994 |
|
Savage CO, Cooke SP: The role of the endothelium in systemic vasculitis. J Autoimmun 6: 237, 1993 |
|
Verani J, Ward PA: Mechanisms of endothelial cell injury in acute inflammation (editorial). Shock 2: 311, 1994 |
|
Bakker WW, Timmerman W, Polestra K et al: Enhanced intraplacental coagulation associated with decreased vascular adp-ase activity in experimental endotoxemia in pregnancy (abstr). Presented at the Rochester Trophoblast Conference, October 1988 |
|
Silen M, Firpo A, Morgello S et al: Interleukin-1 alpha and tumor necrosis factor alpha cause placental injury in the rat. Am J Pathol 135: 239, 1989 |
|
Romero R, Hanaika S, Mazor M et al: Meconium-stained amniotic fluid: A risk factor for microbial invasion of the amniotic cavity. Am J Obstet Gynecol 164: 859, 1991 |
|
Gersell DJ: Chronic villitis, chronic chorioamnionitis and maternal floor infarction. Semin Diagn Pathol 10: 251, 1993 |
|
Fojaco RM, Hensley GT, Moskowitz L: Congenital syphilis and necrotizing funisitis. JAMA 261: 1788, 1989 |
|
Qureshi F, Jacques SM, Reyes MP: Placental histopathology in syphilis. Hum Pathol 24: 779, 1993 |
|
Kaplan C: The placenta and viral infections. Semin Diagn Pathol 10: 222, 1993 |
|
Petignat P, Vial Y, Laurini R: Fetal varicella-herpes zoster syndrome in early pregnancy: Ultrasonograpic and morphological correlation. Prenat Diagn 21: 121, 2001 |
|
Jordan JA, Huff D, DeLoia JA: Placental cellular immune response in woman infected with human parvovirus B19 during pregnancy. Clin Diagn Lab Immunol 8: 288, 2001 |
|
Koi H, Zhang J, Makrigiannakis A, Getsios S: Differential expression of the coxsackievirus and adenovirus receptor regulates adenovirus infection of the placenta. Biol Reprod 64: 1001, 2001 |
|
Fisher S, Genbacev O, Maidji E, Pereira L: Human cyomegalovirus infection of placental cytrophoblasts in vitro and in utero: Implications for transmission and pathogenesis. J Virol 74: 6808, 2000 |
|
Jatczak B, Gejdel E, Pajak J, Podwinska J et al: Study on risk factors for transplacental viral infections; effect of bacterial factors and double viral infections on virus replication in placenta and amniotic membranes. Placenta 22: 360, 2001 |
|
Tkachuk AN, Moorman AM, Poore JA et al: Malaria enhances expression of CC chemokine receptor 5 on placental macrophages. J Infect Dis 183: 967, 2001 |
|
Behbahani H, Popek E, Garcia P et al: Up-regulation of CCR5 expression in the placenta is associated with human immunodeficiency virus-1 vertical transmission. Am J Pathol 157: 1811, 2000 |
|
Ryder RW, Nsa W, Hassig SE et al: Perinatal transmission of the human immunodeficiency virus type 1 to infants of seropositive women in Zaire. N Engl J Med 320: 1637, 1989 |
|
Van Dyke RB, Korber BT, Popek E et al: The Ariel project: A prospective cohort study of maternal-child transmission of human immunodeficiency virus type 1 in the era of maternal antiretroviral therapy. J Infect Dis 179: 319, 1999 |
|
Jauniaux E, Nessman C, Imbert MC et al: Morphological aspects of the placenta in HIV pregnancies. Placenta 9: 633, 1988 |
|
Schwartz DA, Nahmias AJ: Human immunodeficiency virus and the placenta. Current concepts of vertical transmission in relation to other viral agents. Ann Clin Lab Sci 21: 264, 1991 |
|
Popek EJ, Lewis DE, Hammill HA et al: Placental pathology of 100 infants born to HIV+ women (abstr). Placenta 18: A46, 1997 |
|
Reuben J, Lee B-N, Popek EJ: HIV and the placenta. In: Immunol Allergy Clin North Am, HIV Women Pregnancy 18: 371, 1998 |
|
Russell P: Inflammatory lesions of the human placenta. III. The histopathology of villitis of unknown aetiology. Placenta 1: 227, 1980 |
|
Labarrere C, Althabe O: Chronic villitis of unknown aetiology and intrauterine growth-retarded infants of normal and low ponderal index. Placenta 6: 369, 1985 |
|
Labarrere C, Althabe O, Caletti E et al: Deficiency of blocking factors in intrauterine growth retardation and its relationship with chronic villitis. Am J Reprod Immunol Microbiol 10: 14, 1986 |
|
Salafia CM, Parke AL: Placental pathology in systemic lupus erythematosus and the phospholipid antibody syndrome. Rheum Dis Clin North Am 23: 85, 1997 |
|
Khong TY, Staples A, Moore L et al: Observer reliability in assessing villitis of unknown aetiology. J Clin Pathol 46: 208, 1993 |
|
Labarrere C, Faulk WP, McIntyre J: Villitis in normal term human placentae: Frequency of the lesion determined by monoclonal antibody to HLA-DR antigen. J Reprod Immunol 16: 127, 1989 |
|
Khong TY: Expression of MHC class II antigens by placental villi: No relationship with villitis of unknown origin. J Clin Pathol 48: 494, 1995 |
|
Salafia CM, Vintzileos AM, Silberman L et al: Placental pathology of idiopathic intrauterine growth retardation at term. Am J Perinatol 9: 179, 1992 |
|
Bulmer JN, Rasheed RN, Francis N et al: Placental malaria. I. Pathological classification. Histopathology 22: 211, 1993 |
|
Salafia CM, Haynes N, Merluzzi VJ, Rothlein R: The distribution of ICAM-1 within decidua and placenta, and gestational age-associated changes. Pediatr Pathol 11: 381, 1991 |
|
Goldstein J, Braverman M, Salafia CM, Buckley PJ: The phenotype of human placental macrophages and its variation with gestational age. Am J Pathol 133: 648, 1988 |
|
Hasegawa I, Takakuwa K, Adachi S, Kanazawa K: Cytotoxic antibody against trophoblast and lymphocytes present in pregnancy with intrauterine fetal growth retardation and its relation to anti-phospholipid antibody. J Reprod Immunol 17: 127, 1990 |
|
Russell P, Atkinson K, Krishnan L: Recurrent reproductive failure due to severe placental villitis of unknown etiology. J Reprod Med 24: 93, 1980 |
|
Doss BJ, Green MF, Hill J et al: Massive chronic intervillositis associated with recurrent abortions. Hum Pathol 26: 1245, 1995 |
|
Salafia CM, Maier D, Vogel C et al: Placental and decidual histology in spontaneous abortion: Detailed description and correlations with chromosome number. Obstet Gynecol 82: 295, 1993 |
|
Salafia CM, Ernst L, Pezzullo JC et al: The very low birth weight infant: Maternal complications leading to preterm birth, placental lesions, and intrauterine growth. Am J Perinatol 12: 106, 1995 |
|
Erlendsson K, Steinsson K, Johannsson JH, Giersson RT: Relation of antiphospholipid antibody and placental bed inflammatory vascular changes to the outcome of pregnancy in successive pregnancies of 2 women with systemic lupus erythematosus. J Rheumatol 20: 1779, 1993 |
|
Salafia CM, Vintzileos AM, Bantham KF et al: Placental pathologic findings in preterm birth. Am J Obstet Gynecol 165: 934, 1991 |
|
Salafia CM, Pezzullo JC, Lopez-Zeno JA et al: Placental pathology of preterm preeclampsia. Am J Obstet Gynecol 173: 1097, 1995 |
|
Salafia CM, Mill JF: The value of placental pathology in spontaneous prematurity (review). Curr Opin Obstet Gynecol 8: 89, 1996 |
|
Robertson WB, Brosens I, Dixon HG: Uteroplacental vascular pathology. Eur J Obstet Gynecol Reprod Biol 5: 47, 1975 |
|
Naeye RL: Pregnancy hypertension, placental evidences of low uteroplacental blood flow, and spontaneous premature delivery. Hum Pathol 20: 441, 1989 |
|
Arias F, Rodriquez L, Rayne SC, Kraus FT: Maternal placental vasculopathy and infection: Two distinct subgroups among patients with preterm labor and preterm ruptured membranes. Am J Obstet Gynecol 168: 585, 1993 |
|
Friedman SA, de Groot CJ, Taylor RN et al: Plasma cellular fibronectin as a measure of endothelial involvement in preeclampsia and intrauterine growth retardation. Am J Obstet Gynecol 170: 838, 1994 |
|
Salafia CM, Thorp J, Starzyk A: Placental pathology in spontaneous prematurity. The Placenta: Basic Science and Clinical Practice 14: 169, 2000 |
|
Aardema MW, Oosterhof H, Timmer A et al: Uterine artery Doppler flow and uterplacental vascular pathology in normal pregnancies and pregnancies complicated by pre-eclampsia and small for gestational age fetuses. Placenta 22: 405, 2001 |
|
Redman CW, Sargent IL: Placental debris, oxidative stress and pre-eclampsia. Placenta 21: 597, 2000 |
|
Salafia CM, Xenophon J, Lerer T, Silberman L: Fetal growth and placental pathology in maternal hypertensive diseases. Clin Exp Hypertens B 9:27, 1990 |
|
Salafia CM, Minior VK, Pezzullo JC et al: Intrauterine growth retardation in infants of less than 32 weeks' gestational age: Associated placental pathology. Am J Obstet Gynecol 173: 1049, 1995 |
|
DeWolf F, Brosens I, Renaer M: Fetal growth retardation and the maternal arterial supply of the human placenta in the absence of sustained hypertension. Br J Obstet Gynaecol 87: 678, 1980 |
|
Starzyk KA, Salafia CM, Pezzullo JC et al: Quantitative differences in arterial morphometry define the placental bed in preeclampsia. Hum Pathol 28: 353, 1997 |
|
Pijnenborg R, Bland JM, Robertson WB et al: The pattern of interstitial trophoblastic invasion of the myometrium in early pregnancy. Placenta 2: 303, 1981 |
|
Pijnenborg R, Anthony J, Davey DA et al: Placental bed spiral arteries in the hypertensive disorders of pregnancy. Br J Obstet Gynaecol 98: 648, 1991 |
|
Sheppard BL, Bonnar J: Uteroplacental hemostasis in intrauterine fetal growth retardation. Semin Thromb Hemost 25: 443, 1999 |
|
Many A, Hubel CA, Fisher SJ et al: Invasive cytotrophoblasts manifest evidence of oxidative stress in preeclampsia. Am J Pathol 156: 321, 2000 |
|
Bowie EJW: Lipid-related clotting reactions of clinical significance. Arch Pathol Lab Med 116: 1345, 1992 |
|
Madazli R, Budak E, Calay Z, Aksu MF: Correlation between placental bed biopsy findings, vascular cell adhesion molecule and fibronectin levels in pre-eclampsia. Br J Obstet Gynaecol 107: 514, 2000 |
|
Berg K, Roald B, Sande H: High Lp(a) lipoprotein level in maternal serum may interfere with placental circulation and cause fetal growth retardation. Clin Genet 46: 52, 1994 |
|
Meekens JW, Pijnenborg R, Hanssens M et al: Immunohistochemical detection of lipoprotein(a) in the wall of placental bed spiral arteries in normal and severe preeclamptic pregnancies. Placenta 15: 511, 1991 |
|
Salafia CM, Starzyk KA, Lage JM et al: Lipoprotein(a) deposition in the uteroplacental bed and in basal plate uteroplacental arteries: Atherosclerosis-associated processes in normal and complicated pregnancies. Troph Res 11: 377, 1998 |
|
Andrew A, Bulmer JN, Morrison L et al: Subinvolution of the uteroplacental arteries: An immunohistochemical study. Int J Gynecol Pathol 12: 28, 1993 |
|
Ross R: The pathogenesis of atherosclerosis: An update. N Engl J Med 314, 1986 |
|
Kronenberg F, Kronenberg MF, Kiechl S, Trenkwalder et al: Role of lipoprotein(a) and apolipoprotein(a) phenotype in atherogenesis: prospective results from the Bruneck study. Circulation 14: 1154, 1999 |
|
Rabbani LE, Loscalzo J: Recent observations of the role of hemostatic determinants in the development of the atheromatous plaque. Atherosclerosis 105: 1, 1994 |
|
Myers DA, Nathanielsz PW: Biologic basis of term and preterm labor. Clin Perinatol 20: 9, 1993 |
|
Redman CWG: Current topic: Preeclampsia and the placenta. Placenta 12: 301, 1991 |
|
Dekker GA, de Vries JIP, Doelitzsh PM et al: Underlying disorders associated with severe early onset preeclampsia. Am J Obstet Gynecol 173: 1042, 1995 |
|
Harpel PC, Borth W: Fibrin, lipoprotein(a), plasmin interactions: A model linking thrombosis and atherogenesis. Ann NY Acad Sci 233, 1988 |
|
Eskes TK: Clotting disorders and placental abruption: Homocystein: A new risk factor. Eur J Obstet Reprod Biol 95: 206, 2001 |
|
van der Molen EF, Verbruggen B, Novakova I, Eskes TK et al: Hyperhomocysteinemia and other thrombotic risk factors in women with placental vasculopathy. Br J Obstet Gynaecol 107: 785, 2000 |
|
van der Molen EF, Arends GE, Nelen WL et al: A common mutation in the 5,10-methylenetetrahydrofolate reductase gene as a new risk factor for placental vasculopathy. Am J Obstet Gynecol 182: 1258, 2000 |
|
De Falco M, Pollio F, Scaramellino M et al: Homocysteinaemia during pregnancy and placental disease. Clin Exp Obstet Gynecol 27: 188, 2000 |
|
Goddijn-Wessel TA, Wouters MG, van der Molen EF et al: Hyperomocysteinemia: A risk factor for placental abruption or infarction. Eur J Obstet Gynecol Reprod Biol 66: 23, 1996 |
|
Kupferminc MJ, Fait G, Many A, Gordon D et al: Severe preeclampsia and high frequency of genetic thrombophilic mutations. Obstet Gynecol 96: 45, 2000 |
|
Kuperminc MJ, Eldor A, Steinman N, Many A et al: Increased frequency of genetic thrombophilia in women with complications of pregnancy. N Engl J Med 340: 9, 1999 |
|
Mousa HA, Alfirevicl Z: Do placental lesions reflect thrombophilia state in women with adverse pregnancy outcome? Hum Reprod 15: 1830, 2000 |
|
Ray JG, Laskin CA: Folic acid and homocyst(e)ine metabolic defects and the risk of placental abruption, pre-eclampsia and spontaneous pregnancy loss: A systematic review. Placenta 20: 519, 1999 |
|
Alfirevic Z, Mousa HA, Martlew V, Briscoe L: Postnatal screening for thrombophilia in woman with severe pregnancy complications. Obstet Gynecol 97: 753, 2001 |
|
De Stefano V, Casorelli I, Rossi E et al: Interaction between hyperhomocysteinemia and inherited thrombophilic factors in venous thromboembolism. Semin Thromb Hemost 26: 305, 2000 |
|
Reitsma PH: Genetic heterogeneity in hereditary thrombophilia. Haemostasis 30: 1, 2000 |
|
Zoller B, Garcia de Frutous P, Hillarp A, Dahlback B: Thrombophilia as a multigenic disease. Haematologica 84: 59, 1999 |
|
Bienvenu T: Molecular basis of phenotype heterogeneity in cystic fibrosis. Ann Biol Clin (Paris) 55: 113, 1997 |
|
Khong TY, Chambers HM: Alternative methods of sampling placentas for the assessment of uteroplacental vasculature. J Clin Pathol 45: 925, 1992 |
|
Guzman ER, Shen-schwarz S, Benito C et al: The relationship between placental histology and cervical ultrasonography in women at risk for pregnancy losss and spontaneous preterm birth. Am J Obstet Gynecol 181: 793, 1999 |
|
Arias F, Rodrigues L, Rayne SC, Kraus FT: Maternal placental vasculopathy and infection: Two distinct subgroups among patients with preterm labor and preterm ruptured membranes. Am J Obstet Gynecol 168: 585, 1993 |
|
Hansen AR, Collins MH, Genest D et al: Very low birthweight placenta: Clustering of morphologic characteristics. Pediatr Dev Pathol 3: 431, 2000 |
|
Trudinger BJ, Giles WB, Cook CM: Uteroplacental blood flow velocity-time waveforms in normal and complicated pregnancy. Br J Obstet Gynaecol 92: 39, 1985 |
|
Lunell NO, Lewander R, Mamoun I et al: Uteroplacental blood flow in pregnancy-induced hypertension. Scand J Clin Lab Invest 169: 28, 1984 |
|
Sagol S, Ozkinay E, Oztekin K, Ozdemir N: The comparison of uterine artery Doppler velocimetry with the histopathy of the placental bed. Aust NZ J Obstet Gynecol 39: 324, 1999 |
|
Redline RW, Patterson P: Pre-eclampsia is associated with an excess of proliferative immature intermediate trophoblast. Hum Pathol 26: 594, 1995 |
|
Brosens I, Renaer M: On the pathogenesis of placental infarcts in preeclampsia. J Obstet Gynaecol Br Commonw 79: 794, 1972 |
|
Wigglesworth S, Singer DB: Textbook of Fetal and Perinatal Pathology. Cambridge, MA, Blackwell Scientific Publications, 1991 |
|
Mooney EE, Shunnar A, O'Regan M, Gillan JE: Chorionic villous haemorrhage is associated with retroplacental haemorrhage. Br J Obstet Gynaecol 101: 965, 1994 |
|
Batcup G, Tovey LAD, Longster G: Fetomaternal blood group incompatibility studies in placental intervillous thrombosis. Placenta 4: 449, 1983 |
|
Kaplan C, Blanc WA, Elias J: Identification of erythocytes in intervillous thrombi: A study using immunoperoxidase identification of hemoglobins. Hum Pathol 13: 554, 1982 |
|
Harris BA. Peripheral placental separation: A review. Obstet Gynecol 43, 1988. |
|
Giles WB, Trudinger BJ, Baird PJ: Fetal umbilical artery flow velocity waveforms and placental resistance. Pathological correlation. Br J Obstet Gynaecol 92: 31, 1985 |
|
Los FJ, DeWolf BT, Huisjes HJ: Raised maternal serum-alpha fetoprotein levels and spontaneous fetomaternal transfusion. Lancet 1210, 1979 |
|
Giles WB, Trudinger BJ, Baird PJ: Fetal umbilical artery flow velocity waveforms and placental resistance: Pathological correlation. Br J Obstet Gynaecol 92: 31, 1985 |
|
Groome LJ, Owen J, Neely CL et al: Oligohydramnios: Antepartum fetal urine production and intrapartum fetal distress. Am J Obstet Gynecol 165: 1077, 1991 |
|
Bukovsky A, Labarrere CA, Haag B et al: Tissue factor in normal and transplanted human kidneys. Transplant 54: 644, 1992 |
|
Torry RJ, Labarrere CA, Gargiulo P, Faulk WP: Natural anticoagulant and fibrinolytic pathways in renal allograft failure. Transplant 58: 926, 1994 |
|
Silver RK, Peaceman AM, Adams DM: Understanding prostaglandin metabolites and platelet-activating factor in the pathophysiology and treatment of the antiphospholipid syndrome. Clin Perinatol 22: 357, 1995 |
|
Wallenburg HC: Low-dose aspirin therapy in obstetrics. Curr Opin Obstet Gynecol 7: 135, 1995 |
|
Gleicher N, Fribert J: IgM gammopathy and the lupus anticoagulant syndrome in habitual aborters. JAMA 253: 3278, 1985 |
|
Carreras LO, Vermylen J, Spitz B et al: Lupus anticoagulant and inhibition of prostacyclin formation in patients with repeated abortion, intrauterine growth retardation and intrauterine death. Br J Obstet Gynaecol 88: 890, 1981 |
|
Petri M, Golbus M, Anderson R et al: Antinuclear antibody, lupus anticoagulant, and anticardiolipin antibody in women with idiopathic habitual abortion. Arthritis Rheum 30: 601, 1987 |
|
Branch DW, Scott JR, Kochenour KK, Hershgold E: Obstetric complications associated with the lupus anticoagulant. N Engl J Med 313: 1322, 1985 |
|
Triplett A, Harris EN: Antiphospholipid antibodies and reproduction. Am J Reprod Immunol 21: 123, 1989 |
|
DeWolf F, Carreras LO, Moerman P et al: Decidual vasculopathy and extensive placental infarction in a patient with repeated thromboembolic accidents, recurrent fetal loss, and a lupus anticoagulant. Am J Obstet Gynecol 142: 829, 1982 |
|
Feinstein DI: Lupus anticoagulant, thrombosis, and fetal loss. N Engl J Med 21: 1348, 1985 |
|
Cowchock S, Smith JB, Gocial B: Antibodies to phospholipids and nuclear antigens in patients with repeated abortions. Am J Obstet Gynecol 155: 1002, 1986 |
|
Triplett DA: Pathogenic mechanisms of action: Effects on the coagulation system. Fifth International Symposium on Antiphospholipid Antibodies, San Antonio, Texas, 1992 |
|
Griffun JH: Clinical studies of protein C. Semin Thromb Hemost 10: 162, 1984 |
|
Rigby TB, Nolan TE: Inherited disorders of coagulation in pregnancy. Clin Obstet Gynecol 38: 497, 1995 |
|
Khong TY, Sawyer IH, Heryet AR: An immunohistologic study of endothelialization of uteroplacental vessels in human pregnancy. Evidence that endothelium is focally disrupted by trophoblast in preeclampsia. Am J Obstet Gynecol 167: 751, 1992 |
|
Peaceman AM, Rehnberg KA: The effect of aspirin and indomethacin on prostacyclin and thromboxane production by placental tissue incubated with immunoglobulin G fractions from patients with lupus anticoagulant. Am J Obstet Gynecol 173: 1391, 1995 |
|
Hauth JC, Goldenberg RL, Parker CR et al: Maternal serum thromboxane B2 reduction versus pregnancy outcome in a low-dose aspirin trial. Am J Obstet Gynecol 173: 578, 1995 |
|
Morris JM, Fay RA, Ellwood DA et al: A randomized controlled trial of aspirin in patients with abnormal uterine artery blood flow. Obstet Gynecol 87: 74, 1996 |
|
Owen J, Maher JE, Hauth JC et al: The effect of low-dose aspirin on umbilical artery Doppler measurements. Am J Obstet Gynecol 169: 907, 1993 |
|
Sibai BM, Caritis SN, Thom E et al: Prevention of preeclampsia with low-dose aspirin in healthy, nulliparous pregnant women. The National Institute of Child Health and Human Development Network of Maternal-Fetal Medicine Units. N Engl J Med 329: 1213, 1993 |
|
Hamid R, Robson M, Pearce JM: Low-dose aspirin in women with raised maternal serum alpha-fetoprotein and abnormal Doppler waveform patterns from the uteroplacental circulation. Br J Obstet Gynecol 101: 481, 1994 |
|
Hauth JC, Goldenberg RL, Parker CR Jr et al: Low-dose aspirin: Lack of association with an increase in abruptio placentae or perinatal mortality. Obstet Gynecol 85: 1055, 1995 |
|
Cusick W, Salafia CM, Ernst L et al: Low-dose aspirin therapy and placental pathology in women with poor prior pregnancy outcomes. Am J Reprod Immunol 34: 141, 1995 |
|
Viinikka L, Hartikainen-Sorri AL, Lumme R et al: Low-dose aspirin in hypertensive pregnant women: Effect on pregnancy outcome and prostacyclin-thromboxane balance in mother and newborn. Br J Obstet Gynaecol 100: 809, 1993 |
|
Labarrere CA, Faulk WP: Anchoring villi in human placental basal plate: Lymphocytes, macrophages and coagulation. Placenta 12: 173, 1991 |
|
Levy R, Nelson DM. To be, or not to be, that is the question. Apoptosis in human trophoblast. Placenta 21:1, 2000 |
|
Vern TZ, Alles AJ, Kowal-Vern A et al: Frequency of factor V(Leiden) and prothrombin G20210A in placentas and their relationship with placental lesions. Hum Pathol 31: 1036, 2000 |
|
Kraus FT, Acheen VI: Fetal thrombotic vasculopathy in the placenta: cerebral thrombi and infarcts, coagulopathies, and cerebral palsy. Hum Pathol 30: 759, 1999 |
|
Redline RW, Pappin A: Fetal thrombotic vasculopathy: The clinical significance of extensive avascular villi. Hum Pathol 26: 80, 1995 |
|
Demir R, Kaufmann P, Castellucci M et al: Fetal vasculogenesis and angiogenesis in human placental villi. Acta Anat 136: 190, 1989 |
|
Kaufmann P, Luckhardt M, Leiser R: Three-dimensional representation of the fetal vessel system in the human placenta. Trophoblast Res 3: 113, 1988 |
|
McGowan LM, Mullen BM, Ritchie K: Umbilical artery flow velocity waveforms and the placental vascular bed. Am J Obstet Gynecol 157: 900, 1987 |
|
Fok RY, Pavlova Z, Benirschke K et al: The correlation of arterial lesions with umbilical artery Doppler velocimetry in placentas of small-for-dates pregnancies. Obstet Gynecol 75: 578, 1990 |
|
Bracero LA, Beneck D, Kirshenbaum N et al: Doppler velocimetry and placental disease. Am J Obstet Gynecol 161: 388, 1989 |
|
Arabin B, Jimenez E, Vogel M, Weitzel HK: Relationship of utero and fetoplacental blood flow velocity wave forms with pathomorphological placental findings. Fetal Diagn Ther 7: 173, 1992 |
|
Laurini R, Laurini J, Marsal K: Placental histology and fetal blood flow in intrauterine growth retardation. Acta Obstet Gynecol Scand 73: 529, 1994 |
|
Trudinger BJ: Doppler ultrasonography: Applications in clinical practice. In Hanson MA, Spencer JAD, Rodeck CH (eds): The Circulation: Fetus and Neonate, Physiology and Clinical Applications, vol 1. Cambridge, England, Cambridge University Press, 1993 |
|
Rankin JHG, McLaughlin MK: The regulation of placental blood flows. J Dev Physiol 1: 3, 1979 |
|
Salafia CM, Pezzullo JC, Minior VK, Divon MY: Placental pathology of absent and reversed end-diastolic flow in growth restricted fetuses. Obstet Gynecol 90: 830, 1997 |
|
Read MA, Boura ALA, Walters WAW: Effects of variation in oxygen tension on responses of the human fetoplacental vasculature to vasoactive agents in vitro. Placenta 16: 667, 1995 |
|
Macara L, Kingdom JCP, Kohnen G et al: Elaboration of stem villous vessels in growth restricted pregnancies with abnormal umbilical artery Doppler waveforms. Br J Obstet Gynecol 102: 807, 1995 |
|
Hitschold T, Weiss E, Beck T et al: Low target birth weight or growth retardation? Umbilical Doppler flow velocity waveforms and histometric analysis of the feto-placental vascular tree. Am J Obstet Gynecol 168: 1260, 1993 |
|
Jackson MR, Walsh AJ, Morrow RJ et al: Reduced placental villous tree elaboration in small-for-gestational age newborn pregnancies: Relationship with umbilical artery Doppler waveforms. Am J Obstet Gynecol 172: 518, 1995 |
|
Macara L, Kingdon JCP, Kaufmann P et al: Structural analysis of placental terminal villi from growth-restricted pregnancies with abnormal umbilical artery Doppler waveforms. Placenta 17: 37, 1996 |
|
Burton GJ, Jauniaux E: Sonographic, stereological and Doppler flow velocimetric assessments of placental maturity. Br J Obstet Gynecol 102: 818, 1995 |
|
Burton GJ, Reshetnikova OS, Milovanov AP, Teleshova OV: Stereological evaluation of vascular adaptations in human placental villi to differing forms of hypoxic stress. Placenta 17: 49, 1996 |
|
Mak KKW, Gude NM, Walters WAW, Boura ALA: Effects of vasoactive autacoids on the human umbilical fetal placental vasculature. Br J Obstet Gynaecol 91: 99, 1984 |
|
Park HW, Kapur RP, Shepard TH: Reversed circulation in acardiac fetuses is associated with anatomic inversions in the aortic wall. Teratology 49: 267, 1994 |
|
Baldwin VJ: Intertwin vascular anastomoses. In: Pathology of Multiple Pregnancy, p 216. New York, Springer-Verlag, 1994 |
|
Karimu AL, Burton GJ: Significance of changes in fetal perfusion pressure to factors controlling angiogenesis in the human term placenta. J Reprod Fertil 102: 447, 1994 |
|
Karimu AL, Burton GJ: Significance of changes in fetal perfusion pressure to factors controlling angiogenesis in the human term placenta. J Reprod Fertil 102: 447, 1994 |
|
Salafia CM: Fetal and placental pathology in diabetic patients. In Reece EA, Coustan DR (eds): Diabetes Mellitus in Pregnancy: Principles and Practice. New York, Churchill Livingstone, 1988 |
|
Altshuler G: Some placental considerations related to neurodevelopmental and other disorders. J Child Neurol 8: 78, 1993 |
|
Eriksson JG, Forsen T, Tuomilehto J et al: Early growth and coronary heart disease in later life: Longitudinal study. Br Med J 21: 949, 2001 |
|
Law CM, Egger P, Dada O et al: Body size at birth and blood pressure among children in developing countries. Int J Epidemiol 30: 52, 2001 |
|
Eriksson JG, Forsen T, Tuomilehto J et al:. Early growth, adult income, and risk of stroke. Stroke 31: 869, 2000 |
|
Forsen T, Eriksson J, Tuomilehto J et al: The fetal and childhood growth of persons who develop type 2 diabetes. Ann Intern Med 133: 176, 2000 |
|
Shiell AW, Campbell DM, Hall MH, Barker DJ: Diet in late pregnancy and glucose-insulin metabolism of the offspring 40 years later. Br J Obstet Gynecol 107: 890, 2000 |
|
Andersson SW, Bengtsson C, Hallberg L et al: Cancer risk in Swedish women: The relation to size at birth. Br J Cancer 84: 1193, 2001 |
|
Hubinette A, Lichtenstein P, Ekbom A, Cnattingius S: Birth characteristics and breast cancer risk: A study among like-sexed twins. Int J Cancer 91: 248, 2001 |
|
Potischman N, Troisi R. In-utero and early life exposures in relation to risk of breast cancer. Cancer Causes Control 10: 561, 1999 |
|
Burd L, Severud R, Kerbeshian J, Klug MG: Prenatal and perinatal risk factors for autism. J Perinat Med 27: 441, 1999 |
|
Wahlbeck K, Forsen T, Osmond C et al: Association of schizophrenia with low maternal body mass index, small size at birth,and thinness during childhood. Arch Gen Psychiatry 58: 48, 2001 |
|
Conde-Agudelo A: Paternal and maternal components of the predisposition to preeclampsia. N Engl J Med 345: 149, 2001 |
|
Chambers JC, Fusi L, Malik IS et al: Association of maternal endothelial dysfunction with preeclampsia. JAMA 285: 1607, 2001 |
|
Smith GC, Pell JP, Walsh D: Pregnancy complications and maternal risk of ischaemic heart disease: A retrospective cohort study of 129,290 births. Lancet 357: 2002, 2001 |
|
Ros HS, Lichtenstein P, Ekbom A, Cnattingius S: Tall or short? Twenty years after preeclampsia exposure in utero: Comparisons of final height, body mass index, waist-to-hip ratio, and age at menarche among women, exposed and unexposed to preeclampsia during fetal life. Pediatr Res 49: 763, 2001 |