In basic physiologic terms, the placenta is a site of exchange between the maternal and fetal bloodstreams. Nutrients and oxygen are extracted from the maternal circulation, and metabolic byproducts, hormones, and many other molecules are passed into the maternal circulation from the placenta. The placenta transfers many molecules into the fetoplacental pool proportionate to their concentrations in the maternal circulation. The three major modifiers of placental transfer function are maternal blood flow, fetoplacental blood flow, and placental trophoblast membrane permeability. By extension, factors that can limit fetal oxygenation, nutrition, and metabolism are (1) altered maternal perfusion, (2) altered fetoplacental perfusion, (3) reduced placental permeability, and (4) increased placental metabolic needs. Battaglia and Meschia describe the factors determining net fetal nutrient transfer: “… properties of the placenta, such as maternal and fetal blood flows, the pattern of placental perfusion, the surface, thickness and physicochemical properties of the placental membrane, the metabolic activity of the placenta, the various mechanisms of transfer which are available (e.g., diffusion, carrier mediated transfer, active transfer), and regional differences in placental histology and function” (our italics).1 High uterine blood flow, uniform fetal perfusion of the placenta, production of a fetal hemoglobin with greater oxygen affinity than maternal hemoglobin, high fetal cardiac activity, and increased fetal tissue perfusion are the main means by which optimal fetal oxygenation is assured. Normal placental growth and development will maximize placental efficiency while controlling its growth and metabolic needs.
To establish a flourishing intrauterine pregnancy, the trophoblast must anchor to and invade the decidualized endometrium,2 and the uterine vasculature must be able to permit dramatic, progressive increases in blood flow. The implantation process remodels the uterine epithelium and stroma to establish a line of nutrient supply from the maternal tissues and circulation to the conceptus.3 Endometrial invasion has two components, interstitial invasion of the trophoblast into the decidua and stroma, and the endovascular route, by which the vascular remodeling is effected. Interstitial invasion of the endometrium probably involves both an infiltrative and a phagocytic or erosive component.4,5 The infiltrative process involves a complex orchestration of cell adhesion molecule and integrin sequential expressions. The erosive process likely involves far more primitive molecular signals6 and may be linked to the primitive immune mechanisms represented by the abundance of natural killer cells in the normal late luteal and early pregnant human endometrium.7
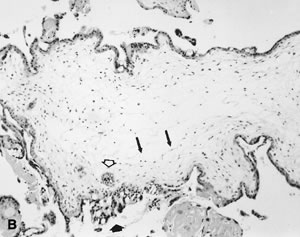
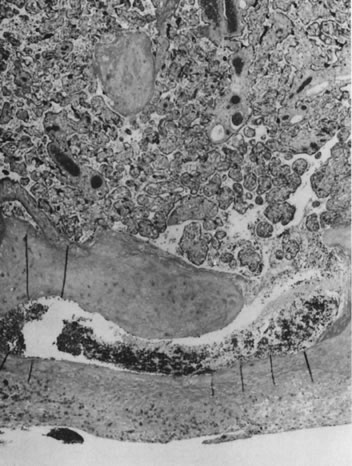
The invasive trophoblast is believed to destroy the nonpregnant uteroplacental spiral arterial wall, replacing the media with a nonmuscular, nonelastic (and likely “pressure-passive”) fibrinoid and temporarily serving as the lining cells to the remodeled arterial structure.8,9,10 Much uterine vascular remodeling occurs without local trophoblast, however (Fig. 1A). Vascular effects may still be the result of secreted trophoblast products, but some aspects of vascular remodeling may require the immediate presence of trophoblasts. Boyd and Hamilton11 describe marked vascular alterations as the spiral arteries “approach the trophoblastic shell,” including marked endothelial hypertrophy disrupting the normal smooth contour of the vessel lumen, and increasing attentuation of the media to the point that the lining endothelium is surrounded only by a layer of reticular or collagen fibers (Fig. 1B). Some of these early effects may be initiated by maternal (rather than fetoplacental) signals. The better understanding of non—trophoblast-dependent(and potentially pre-implantation) uterine vascular changes may permit improved in vitro fertilization success rates, and fewer later gestational complications.12,13
|
The trophoblast's intense vasotropism serves the purpose of establishing, from the earliest days of gestation, a maternal circulation providing the conceptus with nutrients. This circulation may be a sluggish capillary-derived blood pool. The circulation evolves, with spiral arterial remodeling, into a high-volume (capacitance), low-resistance circuit. The repeated failure to identify intervillous circulation both on direct visualization of the intervillous space and using perfused hysterectomy specimens before the 10th to12th week of pregnancy,14,15 combined with documentation of a low-oxygen environment within the coelomic cavity,16 make a standard arterial circulation improbable during embryogenesis. In this schema, too early initiation of maternal arterial perfusion of the intervillous space may damage the conceptus (via oxidative stress)17 and cause early pregnancy loss.18 Others have suggested that fixation artifact and intervillous flow rates (below the current limits of Doppler resolution) may explain most of the flow observations,19 but the evidence for the early embryo's vulnerability to oxidative injury is also compelling.17 Early pregnancy intervillous flow is not likely a brisk arterial one, and is likely part of a highly unusual period of intensive cellular proliferation and differentiation despite comparative hypoxia.
Retrograde trophoblast extension into spiral vessels commences in the venous circuit, and proceeds against the direction of flow into the arterial tree.20 Trophoblastic remodeling is often considered to proceed in two waves, the first completed by the late first trimester. When the second wave is completed, by the early second trimester,21 invasive trophoblast has penetrated the superficial one third of the myometrium. The “waves” of invasion are probably a continuous process, with a more open-ended completion date. Especially in pathologic pregnancy, endovascular trophoblast can be seen in the basal plate level uteroplacental arteries well into the third trimester. Doppler studies have demonstrated a progressive decrease in the downstream resistance in the uterine circulation from implantation,22 which is paralleled by a fall in umbilcal impedance.23 Uterine artery flow increases from 1% to 2% of cardiac output in the nonpregnant state to as much as 30% to 50% over baseline values by 20 to 24 weeks' gestation.24 Complete conversion includes the re-endothelialization of the maternal artery. Failure to complete regrowth of maternal endothelia over the new uteroplacental fibrinoid wall is part of the uteroplacental vascular pathology of preeclampsia.25
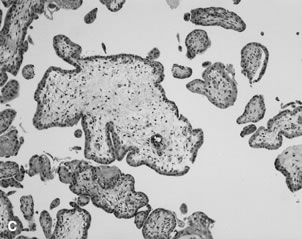
Converted uteroplacental vessels (Fig. 2), 25 no longer responsive to vasomotor stimuli, empty into the intervillous space. Nonpregnant spiral arterial flow is a low-volume (capacitance), high-resistance system. After trophoblast remodeling of spiral vessels, the intervillous space is a high-capacitance, low-resistance system (blood pressure of approximately 10 to 12 mm Hg) that is much larger than apparent in the delivered placenta. In a perfused hysterectomy specimen at the eighth month, the intervillous space occupied half the volume of the placenta.26 In the relaxed (noncontracting) uterus, intervillous pressure is similar to intra-amniotic pressure (at approximately 6 to 10 mm Hg).27
|
The uterine arterial vasculature is comparatively and transiently denervated by pregnancy.28,29 Because the placenta and umbilical cord are not innervated,30 perfusion within the uteroplacental and fetoplacental circulations must depend on anatomic and/or humoral mediators. There is decreased trophoblastic invasion of decidua at the placental margins compared to the center, with less extensive uteroplacental vascular conversion.31 Thus the placental margins are less well perfused, less functional zones that contribute little to placental exchange functions. Routine sampling of placental margins is of limited value.
Three types of trophoblast can be distinguished by their anatomic sites: (1) villous trophoblast, lining the intervillous blood space with cytotrophoblast “stem cells” producing mature syncytiotrophoblast; (2) anchoring (mononuclear) trophoblast that physically anchor villi to the maternal basal plate; and (3) invasive interstitial and endovascular trophoblast that we have already discussed. Interstitial trophoblast cells may fuse (to form multinucleate “placental bed giant cells”) coincidently with the completion of uteroplacental vascular conversion.31,32 Multinucleate giant cells can be seen in the basal plate of pregnancies spontaneously lost in the late first or early second trimester, before vascular conversion should be complete. In these, we question if early trophoblast fusion is a marker of pathologic maternal-placental interaction (and by extension, uteroplacental vascular conversion). All types of trophoblast differentiate and express antigens in highly coordinated and distinct patterns that are influenced by soluble factors and extracellular matrix components.33,34 Villous trophoblast does not express class I or class II histocompatibility antigens. Invasive trophoblasts express human leukocyte antigen G (HLA-G), a major histocompatibility antigen, expression of which is restricted to a few cell types, including trophoblast.35 Recent reviews summarize current data regarding regulation and modulation of trophoblast functions in implnatation and early placental development.36,37,38,39
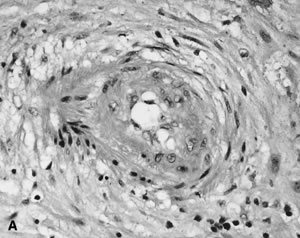
Villous morphologic changes in pregnancy are paralleled by placental functional maturation (Fig. 3A). The “primary villi” are columns of cytotrophoblast that extend from the cytotrophoblastic shell. The intervillous space at this age is essentially labyrinthine. Mesodermal invasion into (or in situ differentiation within) primary villi leads to “secondary villi.” The appearance of blood vessels transforms these into “tertiary villi.”40 The mature villous functional unit is a barrel, with the staves formed by the large fetal stem vessels, and the villous tree arborizing toward the center where the most recently formed villi are found.40 The fetal-neonatal postmaturity syndrome is considered to indicate insufficiency of the aging placenta,41 but some placental growth continues until late in gestation.42 If a placenta older than 42 weeks shows healthy terminal villi, and indication of active villous growth, a cause for fetal/neonatal compromise other than “placental insufficiency” may be sought.
|
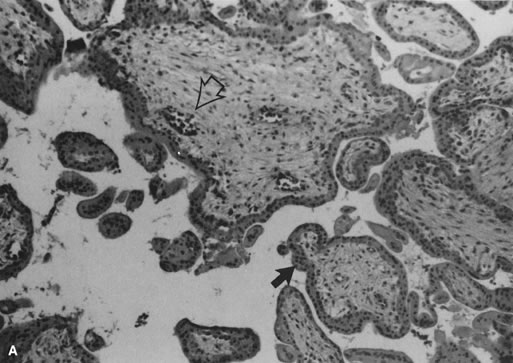
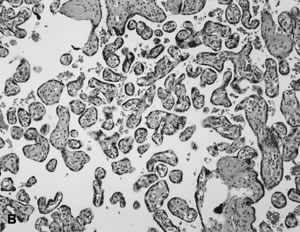
The beating embryonic heart begins to pump nucleated erythrocytes from the yolk sac throughout the villus circulation by the end of the fifth week of gestation. The proportion of nucleated and anucleate erythrocytes in the fetoplacental circulation is tightly correlated with crown-rump length.43 Villous macrophages (Hofbauer cells) are numerous throughout gestation (40% of stromal cells),44 although at term they may be less conspicuous. Villi arborize by mesenchymal invasion of syncytial sprouts.45 Sprouts can be deported into the maternal circulation, where they may influence maternal tolerance of the conceptus. Villi originally develop over the entire sphere of the conceptus, but atrophy over the extraplacental membranes by the end of the first trimester. Villous atrophy and tissue death related to this normal part of placental development may be difficult to distinguish from pathologic villous necrosis (e.g., infarcts). Remnants of ghost villi can be seen on the free membranes (chorion laeve) even at term. The chorion frondosum develops into the placental disc. Aberrant persistence of proliferating villi distant from the main placenta disk form “accessory” or succenturiate lobes.
The delivered placenta is “deflated” compared to its state in utero, a consideration for anyone attempting ultrasound or anatomic correlations.26,46 The average diameter of the delivered placenta at term has been reported as 18.5 cm (range 10.5 to 24.5 cm), with a mean thickness of 2.3 cm (range 1.1 to 4.1 cm).46 The same source described the early growth of the placenta as primarily an increase in diameter of the chorionic disc, while in the later stages of gestation, placental weight increased primarily due to an increase in placental thickness.46 They likewise reported the mean diameters of the placenta at the third to sixth months as 5.8 cm, 8.2 cm, 10.8 cm, and 13.0 cm, respectively. The number of major villous trunks remains constant;45 therefore, placental growth requires that each major trunk (established in the early months of gestation) must arborize or otherwise develop proportional to the increase in placental weight from early gestation to delivery. Schneider has recently reviewed the ultrasound data regarding placental volume in the midtrimester.47 The mean weekly increase in placental volume between 16 and 24 weeks was 31, ±, 8 cm3; further, placental volume in the second trimester was closely correlated with infant birthweight, suggesting that midtrimester placental well-being is an important determiner of late fetal growth.
Early villous vessels are situated deep in the villous stroma, at a mean distance of 24 to 27 μm from the intervillous space to villous vessel lumen (Fig. 3A).45 Placental mass increases more slowly than fetal weight by the end of the second trimester(Table 1).48 In the third trimester, fetal growth is able to continue disproportionate to the mass of the nurturing placenta because the later placenta has increased diffusion efficiency. Progressive reduction in trophoblast thickness, villous size, and the distance between the intervillous space and the fetoplacental capillary are the anatomic bases for improved transfer efficiency (Fig. 3B). The placental vasculature also increases in complexity, with increasing numbers of capillary outlines per villus, until about 36 weeks' gestation. Additionally, there is a massive enlargement of the syncytiotrophoblastic surface, disproportionate to syncytial volume. Microvillous surface increases until about 36 weeks' gestation. Overall, this increases the villous surface area of exchange.49,50,51
Table 1. Expected Fetal and Placental Weight Ratio
Weeks Gestation | Placental Wt (g) | Fetal Wt (g) | F:P Ratio |
8 | 6 | 1.1 | 0.18 |
10 | 14 | 5 | 0.36 |
12 | 26 | 17 | 0.65 |
14 | 42 | 30 | 0.71 |
16 | 65 | 60 | 0.92 |
18 | 90 | 130 | 1.44 |
20 | 115 | 250 | 2.17 |
22 | 150 | 400 | 2.67 |
24 | 185 | 560 | 3.03 |
26 | 210 | 750 | 3.57 |
28 | 250 | 1000 | 4.00 |
30 | 285 | 1260 | 4.42 |
32 | 315 | 1550 | 4.92 |
34 | 355 | 1900 | 5.35 |
36 | 390 | 2300 | 5.90 |
38 | 425 | 2750 | 6.47 |
40 | 470 | 3400 | 7.23 |
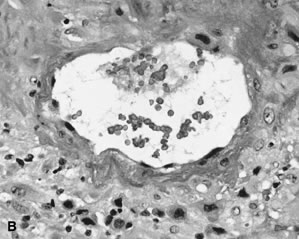
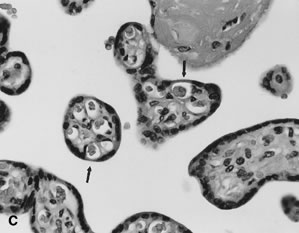
Placental permeability, the ease of transfer across the placental membrane, increases even after placental growth rate peaks by 30 to 32 weeks' gestation.1 This is believed to be due to the formation of “vasculosyncytial membranes.” In a vasculosyncytial membrane, villous capillaries abut the trophoblast basement membrane, and the trophoblast nuclei and cytoplasm are clumped at the edge of the capillary margin (Fig. 3C). The membrane itself measures only 0.5 to 1.0 μ in thickness.45 Mean diffusion distance between the mother's bloodstream and the fetal circulation is ~3 μm.50 Progressive improvement in placental permeability leads to a “flow-limited” placental transfer capacity at term.1
In vivo studies in humans have identified a decrease in the volume of umbilical venous flow in the third trimester.52 Therefore, despite the remodelling of the placenta for increased functional capacity, there is a real potential for decreased fetal supply during the last trimester. At term the weight-specific placental oxygen consumption is almost five times as high as that of the fetus.53 Shortly before birth, the fetus also begins to prepare for extrauterine life by switching synthesis from hemoglobin F (HbF) to hemoglobin A (HbA). Although HbF has a higher oxygen affinity than HbA, which confers an “advantage” to the fetus favoring fetal oxygen extraction from maternal intervillous blood, late in pregnancy this advantage is reduced. These features combine to create a physiologic basis for “placental insufficiency” late in pregnancy and post term as fetal and placental growth, fetal and placental metabolic needs, and the maturational changes in Hb combine potentially at the upper limits of uterine supply.
Placental physiology is not the primary focus of this chapter. Brief highlights of specific aspects of placental physiology are presented here concerning maternal fetal placenta water balance and placenta respiratory function. The placenta is the major determiner of fetal water balance,54 with a relatively insignificant contribution of the fetal kidney. Maternal dehydration is directly paralleled by increased fetal plasma osmolality.55 However, the composition of fetal urine is significantly different from that of fetal plasma, indicating a fetal renal role in ion homeostasis. In the absence of fetal stress, fetal urine (and amniotic fluid) is generally hypotonic.56 Amniotic fluid composition is changed from that of fetal urine as it leaves the kidney via gestational age-dependent changes in extraplacental membrane permeability.56
Oxygen and carbon dioxide are exchanged across the placenta by simple diffusion. Longo has summarized the major factors affecting oxygen transfer.57 The placenta is a very metabolically active tissue, consuming one third to one half of the oxygen presented to the intrauterine tissues.1 Oxygen exchange almost always is flow limited, rather than diffusion limited.57 The fetus may direct placentally supplied nutrients, including oxygen, to different tissue uses at different gestational ages.1 Until 70 torr, maternal arterial partial pressure of oxygen (PO2) has little effect on placental oxygen exchanged; below this value, progressively less oxygen crosses to the fetus. Umbilical venous oxygen tension is generally 10 to 15 torr less than uterine venous blood values. Fetal carbon dioxide production is about equal to PO2 utilization. Fetal carbon dioxide rapidly diffuses into maternal blood depending on the concentration gradient between fetal and maternal blood. Fetal partial pressure of carbon dioxide (PCO2) drops with maternal hyperventilation and rises with maternal hypoventilation, impaired placental perfusion, or permeability.1 Fetal arterial PCO2 can rise during fetal underperfusion of the placenta (or cord compression).1
Carrier-mediated transfer of many molecules across the placenta (including glucose and amino acids) is affected by changes in either maternal or fetal perfusion. The placenta metabolizes most of the glucose obtained from the maternal circulation, and provides the fetus with less moles of glucose than required to fuel the fetus.1 Placental lactate is a major fuel of fetal metabolism. In fetal growth restriction (FGR), fetal nutrient utilization may decrease,58 leading to underutilization of placental lactate and a lactic acidosis. Under conditions of asphyxia, the fetus itself is a net producer of lactate.1 Ketoacids, such as produced in maternal diabetic ketoacidosis, may reduce fetal brain metabolism.1