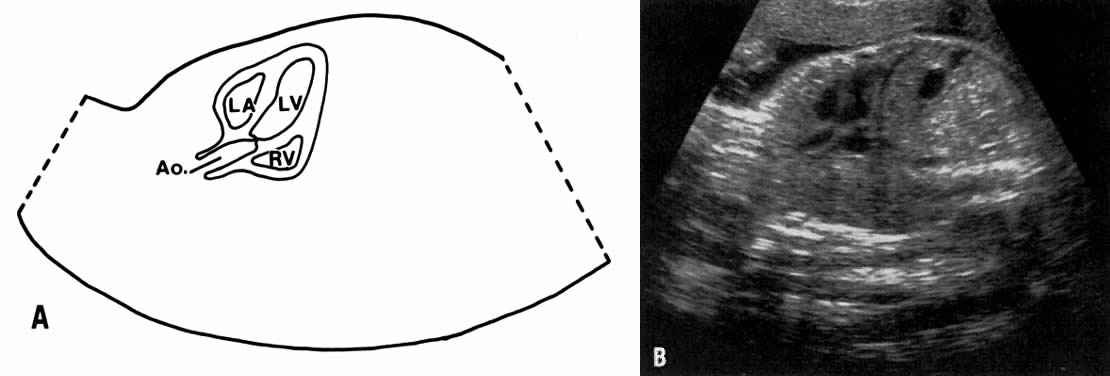
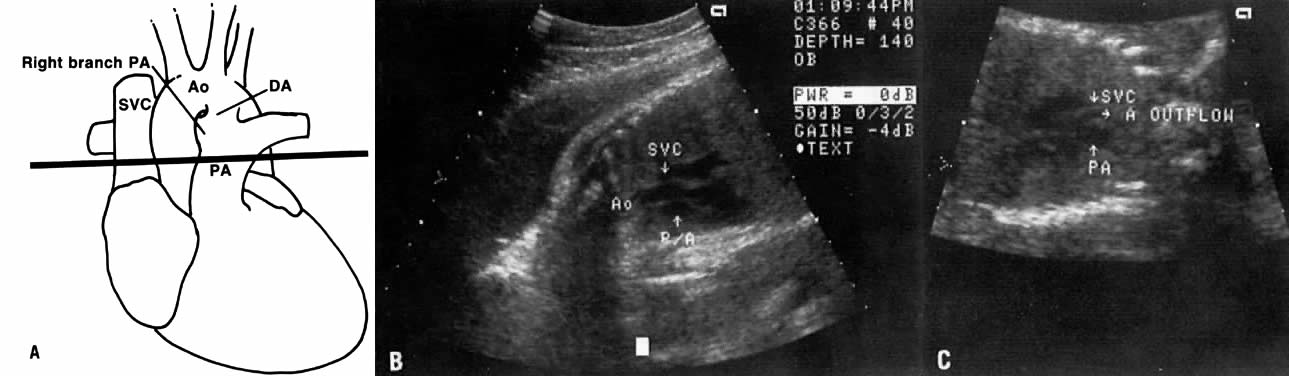
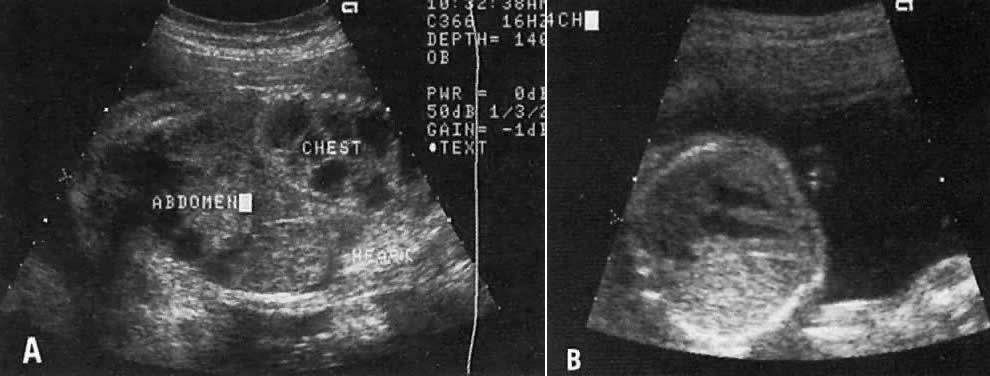
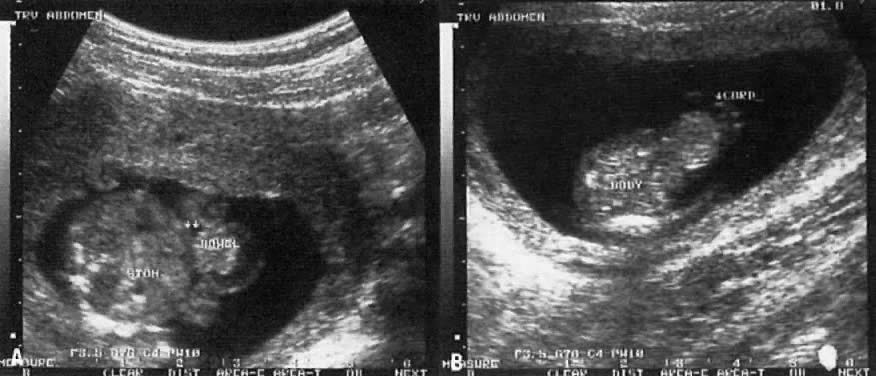
The recent development of high-resolution ultrasound equipment has markedly improved the diagnostic accuracy of ultrasound. In particular, the introduction of high-frequency vaginal probes has enabled early diagnosis of certain fetal abnormalities from the 12th to 14th week of pregnancy. Such early testing is of special importance for women with a history of pregnancies associated with birth defects. The frequency with which different organ systems can be imaged is shown in Table 1.1 The sequential appearance of fetal neural structures during the first trimester of pregnancy is shown in Table 2.2
Gestation (weeks) | <10 | 10–12 | 12–14 | >14 |
Patients | 19 | 34 | 65 | 26 |
Four chamber | - | 13 (37) | 44 (68) | 21 (81) |
Head | - | 13 (37) | 43 (66) | 16 (62) |
Spine | - | 5 (15) | 28 (43) | 8 (31) |
Stomach | 2 (10) | 19 (56) | 62 (95) | 24 (92) |
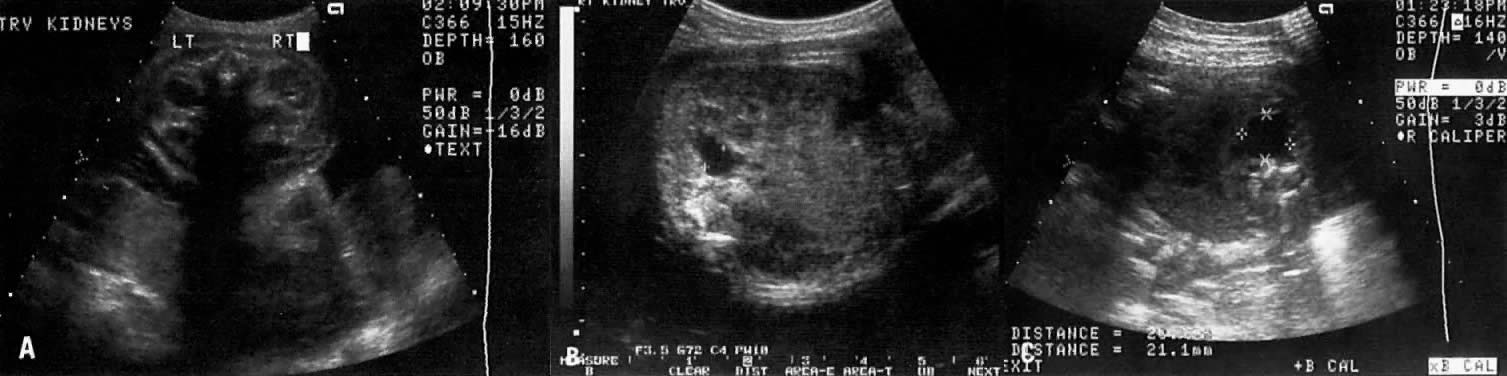
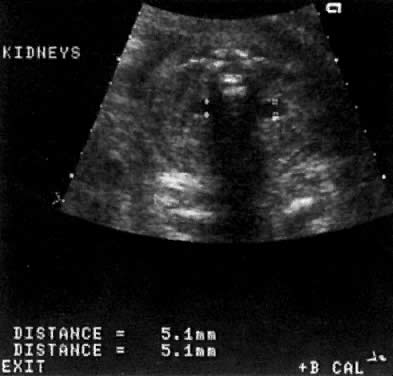
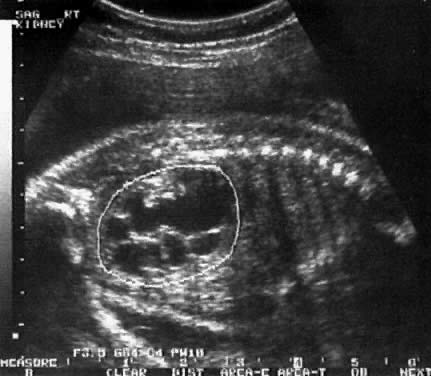
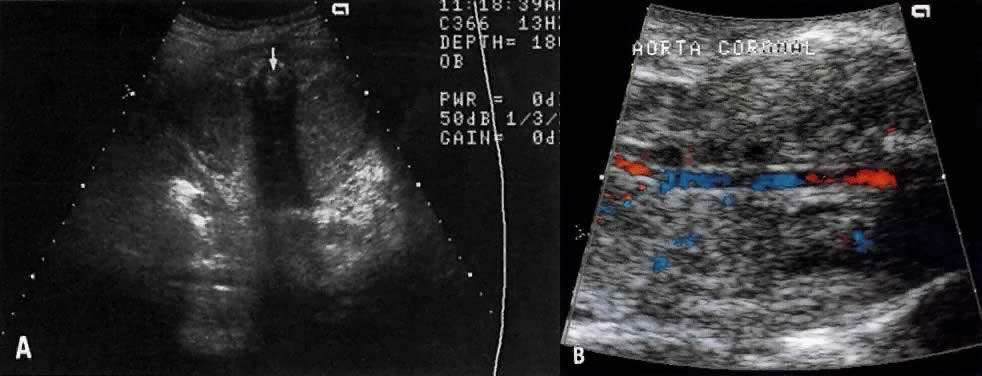
Kidneys | - | 5 (15) | 38 (58) | 14 (54) |
Bladder | 3 (6) | 14 (42) | 52 (80) | 23 (88) |
*Percentages are shown in parentheses. Study included 150 low-risk pregnancies and utilized Acuson equipment and a 5-MHz transducer.
(Adapted from Johnson P, Sharland G. Maxwell D, Allan L: The role of transvaginal sonography in the early detection of congenital heart disease. Ultrasound Obstet Gynecol 2:2481, 1992).
TABLE 2. Sequential Appearance of Fetal Neural Structures During the First
Trimester of Pregnancy
| Mentstrual Age (Weeks) | |||||||
Structure | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 |
Cephalic pole |
| ________________________________→ |
|
| ||||
Univentricular system |
| __________________→ |
|
|
| |||
Falx cerebri |
|
|
| ____________________→ |
| |||
Biventricular system |
|
|
| ______________________→ |
| |||
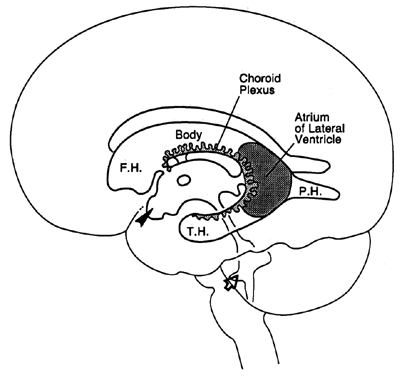
Choroid plexus |
|
|
| ______________________→ |
| |||
Thalamus |
|
|
|
|
| ___________→ |
| |
Third Ventricle |
|
|
|
|
| ___________→ |
| |
Corpus callosum |
|
|
|
|
| ___________→ |
| |
Cerebral peduncles |
|
|
|
|
| ___________→ |
| |
Pons |
|
|
|
|
|
| ____→ | |
Cerebellum |
|
|
|
|
|
| ____→ | |
Cerebellar tentorium |
|
|
|
|
|
| ____→ | |
Hippocampus |
|
|
|
|
|
| ____→ | |
Posterior fossa (cisterna magna) |
|
|
|
|
|
| ____→ | |
IV ventricle |
|
|
|
|
|
| ____→ | |
Cerebral arteries |
|
|
|
|
| ______→ |
| |
Corpus striatum |
|
|
|
|
|
| ____→ | |
Calvarial skeleton |
|
|
|
|
|
| ____→ | |
Spinal skeleton |
|
|
|
|
|
| _→ | |
The examination for the detection of congenital anomalies is referred to as either detailed ultrasound study or targeted imaging for fetal anomalies (TIFFA)3, 4 In such examinations, a variety of fetal anatomic views (“targets”') are specifically sought after and imaged by experienced ultrasonographers. The indications for TIFFA are listed in Table 3.
TABLE 3. Targeted Imaging for Fetal Anomalies: Indications
Poor obstetric history
History of previous affected fetus: multifactorial, genetic or chromosomal
abnormality
Findings on standard obstetric study that increases risk for aneuploidy: choroid
plexus cyst, pyelectases, hyperechogenic bowel, two vessel umbilical
cord, heart defect, cleft lip, increased nuchal thickness, cystic
hygroma, hydrops, diaphragmatic hernia, omphalocele, short humerus
or femur bone.
Findings on standard obstetric study that increase risk for spina bifida: lemon/banana
sign.
Inability to obtain a specific anatomic view on standard study.
Abnormal level of maternal serum or amniotic fluid volume alpha-fetoprotein.
Abnormal maternal triple-screen risk.
Amniotic fluid volume abnormalities.
Intrauterine growth restriction.
Breech at term.
Maternal diabetes mellitus.
Advanced maternal age.
History of adoption.
Exposure to teratogen.
Absent end diastolic velocity by umbilical Doppler study.
Other indications (maternal anxiety, thick placenta)
Despite these technical advances, ultrasound accuracy remains dependent on gestational age. For example, if only one ultrasound examination is to be performed in the second trimester of pregnancy, the best time would be between weeks 19 and 20. Accuracy also depends on the skill of the ultrasonographer in obtaining the correct anatomic views and in differentiating between normal and abnormal structures.
The ultrasonographer also plays an essential role in 1) interpretation of the pathophysiology of a discovered defect, and 2) delineating the risk involved. For example, the risk for spina bifida in a woman with elevated maternal serum alpha-fetoprotein is much lower if a detailed ultrasound study is normal (Table 4).5 Thus, the need for amniocentesis to evaluate amniotic fluid alpha-fetoprotein (which is a better predictor of spina bifida than maternal serum alpha-fetoprotein) should be reevaluated in relation to the newly assigned risk (see Table 4).5 Similarly, the risk for autosomal trisomy is lower than the age-related or maternal triple-screen risk in women with a normal second trimester fetal ultrasound examination (Table 5, Table 6, Table 7 and Table 8.)
AFP | Ventral | Prior Incidence of Open Spina Bifida per 1000 | ||||||||
Multiples | Wall Defect |
| ||||||||
of Median | Only | 0.1 | 0.2 | 0.4 | 0.8 | 1 | 2 | 3.5 | 5 | 25 |
2 | 3600 | 18000 | 9000 | 4500 | 2300 | 1800 | 900 | 510 | 360 | 71 |
| 16000 | 79000 | 39000 | 20000 | 9800 | 7900 | 3900 | 2200 | 1600 | 310 |
| 69000 | 340000 | 170000 | 86000 | 43000 | 34000 | 17000 | 9800 | 6800 | 1300 |
2.5 | 1500 | 4700 | 2400 | 1200 | 590 | 470 | 240 | 140 | 95 | 19 |
| 6300 | 21000 | 10000 | 5100 | 2600 | 2100 | 1000 | 590 | 410 | 81 |
| 28000 | 90000 | 45000 | 22000 | 11000 | 9000 | 4500 | 2600 | 1800 | 350 |
3 | 590 | 1500 | 740 | 370 | 190 | 150 | 75 | 43 | 31 | 6.8 |
| 2600 | 6500 | 3200 | 1600 | 810 | 650 | 320 | 190 | 130 | 26 |
| 11000 | 28000 | 14000 | 7000 | 3500 | 2800 | 1400 | 800 | 560 | 110 |
3.5 | 250 | 530 | 270 | 130 | 68 | 54 | 28 | 16 | 12 | 3.1 |
| 1100 | 2300 | 1200 | 580 | 290 | 230 | 120 | 67 | 47 | 10 |
| 4700 | 10000 | 5100 | 2500 | 1300 | 1000 | 510 | 290 | 200 | 41 |
4 | 110 | 210 | 110 | 54 | 28 | 22 | 12 | 7.1 | 5.2 | 1.8 |
| 470 | 930 | 460 | 230 | 120 | 94 | 47 | 27 | 19 | 4.6 |
| 2000 | 4000 | 2000 | 1000 | 510 | 400 | 200 | 120 | 81 | 17 |
4.5 | 50 | 93 | 47 | 24 | 13 | 10 | 5.6 | 3.6 | 2.8 | 1.4 |
| 210 | 400 | 200 | 100 | 51 | 41 | 21 | 12 | 9 | 2.6 |
| 930 | 1800 | 880 | 440 | 220 | 180 | 88 | 51 | 36 | 7.8 |
5 | 24 | 44 | 22 | 12 | 6.3 | 5.3 | 3.1 | 2.2 | 1.9 | 1.2 |
| 100 | 190 | 94 | 48 | 24 | 20 | 10 | 6.3 | 4.7 | 1.7 |
| 440 | 810 | 410 | 200 | 100 | 82 | 42 | 24 | 17 | 4.2 |
*Each number in the table is the denominator of the risk with a standard numerator of 1.
The left-hand column gives risk of ventral wall defects, and the remaining columns give risk of open spina bifida based on prior population incidence. In each column, the upper figure provides the risk based on prior incidence and on MSAFP.
The lower figure in each column is boldface and is the best estimate of risk combining prior population incidence, MSAFP, and normal ultrasound.
MSAFP = maternal serum alpha-fetoprotein.
(Thornton JG, Lilford RJ, Newcombe RG: Tables for estimation of individual risks of fetal neural tube and ventral wall defects, incorporating prior probability, maternal serum alpha-fetoprotein, and ultrasonographic examination results. Am J Obstet Gynecol 164:154, 1991)
TABLE 5. Ultrasound Findings Associated With Increased Risk For Aneuploidy
Women Younger than 33 Years of Age | Women Older than 33 Years of Age |
(An Abnormal Score is | (An Abnormal Score is |
2 Nuchal Fold | 2 Nuchal fold |
2 Cardiac, CNS or other anatomic defect | 2 Cardiac, CNS or other 2 anatomic defect |
1 Short FL/expected FL* | 1 Short FL/expected FL* |
1 Short HL/expected JL † | 1 Short HL/expected JL † |
1 Pyelectasis ‡ | 1 Pyelectasis ‡ |
| 1 Echogenic bowel |
| 1 Choroid plexus cyst |
Abnormal scores in both age groups increase risk for autosomal trisomy to a level greater than 1:182-the standard age related risk of a 35-year-old pregnant woman at 16 weeks' gestation. As a result, an abnormal score warrants counseling for possible assessment of fetal karyotype. Sensitivity and specificity of abnormal score are > 80%. Similarly, a normal score of 0 after 33 years of age would decrease the risk for aneuploidy (see Table 6).
*Expected femur length (mm) = -9.3105 + 0.9028 × biparietal diameter, value abnormal if measured/expected length .
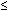 0.9.
0.9.†Expected humeral length (mm) = -7.904 + 0.849 × biparietal diameter, value abnormal if measured/expected length < 0.9.
‡Anteroposterior diameter of renal pelvis
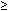 4 mm.
4 mm.(Adapted from Benacerraf BR, Neuberg D, Bromley et al: Sonographic scoring index for prenatal detection of chromosomal abnormalities. J Ultrasound Med 11:449,1992).
|
| After Scan | Lower Limit† |
Maternal Age | Before Scan | (Score = 0) | 95% CI |
33 | 1 : 294 | 1 : 1666 | 0.9 |
34 | 1 : 232 | 1 : 1428 | 1.2 |
35 | 1 : 182 | 1 : 1111 | 1.5 |
36 | 1 : 140 | 1 : 833 | 2.0 |
37 | 1 : 112 | 1 : 666 | 2.5 |
38 | 1 : 87 | 1 : 536 | 3.2 |
39 | 1 : 68 | 1 : 400 | 4.1 |
40 | 1 : 53 | 1 : 312 | 5.3 |
41 | 1 : 42 | 1 : 244 | 6.8 |
42 | 1 : 33 | 1 : 192 | 8.7 |
44 | 1 : 20 | 1 : 116 | 14.5 |
46 | 1 : 12 | 1 : 68 | 24.3 |
*Score of 0 = normal ultrasound findings. Note that a normal score of 0 after 33 years of age would decrease the risk of aneuploidy necessitating reevaluation of need for assessment of fetal karyotype.
† Lower limit of the Confidence Interval for sensitivity and specificity.
(Adapted from Nadel AS, Bromley B, Frigoletto FD JR, Benacerraf BR: Can the presumed risk of autosomal trisomy be decreased in fetuses of older women following a normal sonogram? J Ultrasound Med 14:297, 1995).
|
|
|
|
|
| Short | Pyelectasis |
|
|
|
|
|
| Short | Short | Short | Femur and | with Short |
|
|
|
Maternal | Age or Triple | Normal | Femur | Femurby | Humerus | Humerusby | Femur | Nuchal | Echogeni |
|
Age (yr) | Screen Risk | Ultrasonography | byBPD/FL | O/E | by O/E | O/E | orHumerus | Fold | cBowel | ShortEar |
20 | 1231 | 18,871 | 260 | 463 | 301 | 108 | 194 | 29 | 91 | 194 |
21 | 1145 | 17,553 | 242 | 431 | 280 | 100 | 181 | 27 | 85 | 180 |
22 | 1065 | 16,326 | 225 | 401 | 261 | 93 | 168 | 26 | 79 | 168 |
23 | 1000 | 15,330 | 212 | 376 | 245 | 88 | 158 | 24 | 74 | 157 |
24 | 942 | 14,441 | 199 | 354 | 231 | 83 | 149 | 23 | 70 | 148 |
25 | 887 | 14,598 | 188 | 334 | 217 | 78 | 140 | 21 | 66 | 140 |
26 | 842 | 12,908 | 178 | 317 | 206 | 74 | 133 | 20 | 63 | 133 |
27 | 798 | 12,233 | 169 | 300 | 196 | 70 | 126 | 19 | 59 | 126 |
28 | 755 | 11,574 | 160 | 287 | 185 | 66 | 120 | 18 | 56 | 119 |
29 | 721 | 11,053 | 153 | 271 | 177 | 64 | 114 | 18 | 54 | 114 |
30 | 685 | 10,501 | 145 | 258 | 168 | 60 | 109 | 17 | 51 | 108 |
31 | 650 | 9,964 | 138 | 245 | 159 | 57 | 103 | 16 | 49 | 103 |
32 | 563 | 8,631 | 120 | 212 | 138 | 50 | 89 | 14 | 42 | 89 |
33 | 452 | 6,929 | 96 | 170 | 111 | 40 | 72 | 11 | 34 | 72 |
34 | 352 | 5,396 | 75 | 133 | 87 | 31 | 56 | 9 | 27 | 56 |
35 | 274 | 4,200 | 59 | 104 | 68 | 25 | 44 | 7 | 21 | 44 |
36 | 213 | 3,265 | 46 | 81 | 53 | 19 | 34 | 6 | 17 | 34 |
37 | 166 | 2,545 | 36 | 63 | 41 | 15 | 27 | 5 | 13 | 27 |
38 | 129 | 1,978 | 28 | 49 | 32 | 12 | 21 | 4 | 10 | 21 |
39 | 100 | 1,533 | 22 | 38 | 25 | 10 | 17 | 3 | 8 | 17 |
40 | 78 | 1,196 | 17 | 30 | 20 | 8 | 13 | 3 | 7 | 13 |
41 | 61 | 935 | 14 | 24 | 16 | 6 | 10 | 2 | 5 | 10 |
42 | 47 | 721 | 11 | 18 | 12 | 5 | 8 | 2 | 4 | 8 |
43 | 37 | 567 | 9 | 15 | 10 | 4 | 7 | 2 | 4 | 7 |
44 | 29 | 445 | 7 | 12 | 8 | 3 | 5 | 2 | 3 | 5 |
45 | 22 | 337 | 5 | 9 | 6 | 3 | 4 | 1 | 3 | 4 |
46 | 17 | 261 | 4 | 7 | 5 | 2 | 4 | 1 | 2 | 4 |
47 | 13 | 199 | 4 | 6 | 4 | 2 | 3 | 1 | 2 | 3 |
48 | 10 | 153 | 3 | 4 | 3 | 2 | 2 | 1 | 2 | 2 |
49 | 8 | 123 | 3 | 4 | 3 | 2 | 2 | 1 | 2 | 2 |
FL, femur length: O, observed; E, expected (mean).
Note, as an example, that at maternal age of 36 years the risk for Down Syndrome is 1 : 213. However, following a normal second trimester scan the risk can be adjusted to 1 : 3265. As a result assessment of fetal karyotype would become unnecessary.
(Adapted from Vintzileos AM, Egan JFX: Adjusting the risk for trisomy 21 on the basis of second trimester ultrasonography. Am J Obstet Gynecol 172:837, 1995).
|
|
|
|
|
| Pyelectasis |
|
|
|
Triple |
|
|
| Short | Short Femur | with Short |
|
|
|
Screen | Normal | Short Femur | Short Femur | Humerus | and Humerus | Femur | Nuchal | Echogeni |
|
Risk | Ultrasonography | by BPD/FL | by O/E | by O/E | by O/E | orHumerus | Fold | c Bowel | Short Ear |
15,000 | 160,000 | 3163 | 5631 | 3661 | 1303 | 2359 | 347 | 1100 | 2348 |
14,500 | 154,667 | 3057 | 5444 | 3539 | 1260 | 2281 | 335 | 1063 | 2270 |
14,000 | 149,333 | 2952 | 5256 | 3417 | 1216 | 2202 | 324 | 1026 | 2192 |
13,500 | 144,000 | 2846 | 5068 | 3295 | 1173 | 2124 | 312 | 990 | 2114 |
13,000 | 138,667 | 2741 | 4881 | 3173 | 1129 | 2045 | 300 | 953 | 2035 |
12,500 | 133,333 | 2636 | 2693 | 3051 | 1086 | 1966 | 289 | 917 | 1957 |
12,000 | 128,000 | 2530 | 4505 | 2929 | 1043 | 1888 | 277 | 880 | 1879 |
11,500 | 122,667 | 2425 | 4318 | 2807 | 999 | 1809 | 266 | 843 | 1801 |
11,000 | 117,333 | 2319 | 4130 | 2685 | 956 | 1730 | 254 | 807 | 1722 |
10,500 | 112,000 | 2214 | 3942 | 2563 | 912 | 1652 | 243 | 770 | 1644 |
10,000 | 106,667 | 2109 | 3754 | 2441 | 869 | 1573 | 231 | 733 | 1566 |
9,500 | 101,333 | 2003 | 3567 | 2319 | 826 | 1495 | 220 | 697 | 1488 |
9,000 | 96,000 | 1898 | 3379 | 2197 | 782 | 1416 | 208 | 660 | 1409 |
8,500 | 90,667 | 1792 | 3191 | 2075 | 739 | 1337 | 197 | 624 | 1331 |
8,000 | 85,333 | 1687 | 3004 | 1953 | 695 | 1259 | 185 | 587 | 1253 |
7,500 | 80,000 | 1582 | 2816 | 1831 | 652 | 1180 | 174 | 550 | 1175 |
7,000 | 74,667 | 1476 | 2628 | 1709 | 609 | 1102 | 162 | 514 | 1096 |
6,500 | 69,333 | 1371 | 2441 | 1587 | 565 | 1023 | 151 | 477 | 1018 |
6,000 | 64,000 | 1266 | 2253 | 1465 | 522 | 944 | 139 | 440 | 940 |
5,500 | 58,667 | 1160 | 2065 | 1343 | 478 | 866 | 128 | 404 | 862 |
5,000 | 53,333 | 1055 | 1878 | 1221 | 435 | 787 | 116 | 367 | 783 |
4,500 | 48,000 | 949 | 1690 | 1099 | 392 | 708 | 135 | 331 | 705 |
4,000 | 42,667 | 844 | 1502 | 977 | 348 | 630 | 93 | 294 | 627 |
3,500 | 37,333 | 739 | 1315 | 855 | 305 | 551 | 82 | 257 | 549 |
3,000 | 32,000 | 633 | 1127 | 733 | 261 | 473 | 70 | 221 | 470 |
2,500 | 26,667 | 528 | 939 | 611 | 218 | 394 | 59 | 184 | 392 |
2,000 | 21,333 | 422 | 751 | 489 | 175 | 315 | 47 | 147 | 314 |
1,500 | 16,000 | 317 | 564 | 367 | 131 | 237 | 36 | 111 | 236 |
1.000 | 10,667 | 212 | 376 | 245 | 88 | 158 | 24 | 74 | 157 |
500 | 5,333 | 106 | 188 | 123 | 44 | 80 | 13 | 38 | 79 |
FL, Femur length; O, observed; E, expected (mean)
Note: The finding of short femoral and humeral lengths, relative to expected values (see Table 5), would adjust a triple screen risk for Down syndrome from 1 : 2000 to 1 : 175. This adjustment suggests that further testing to assess fetal karyotype would now be warranted.
(Adapted from Vintzileos AM, Egan JFX: Adjusting the risk for trisomy 21 on the basis of second trimester ultrasonography. Am J Obstet Gynecol 172:837, 1995)
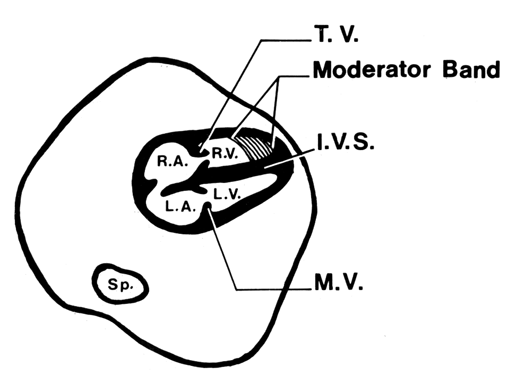
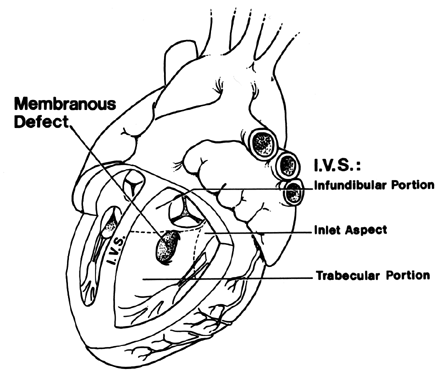
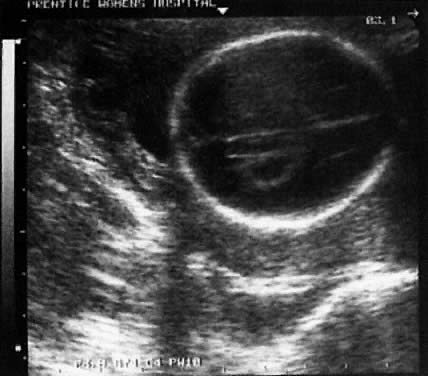
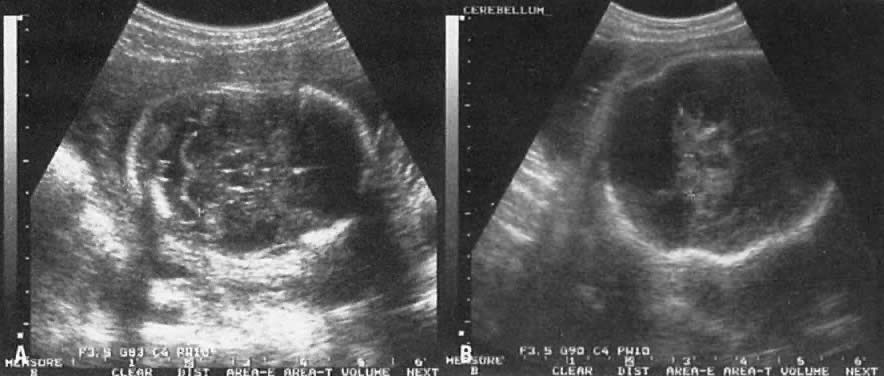
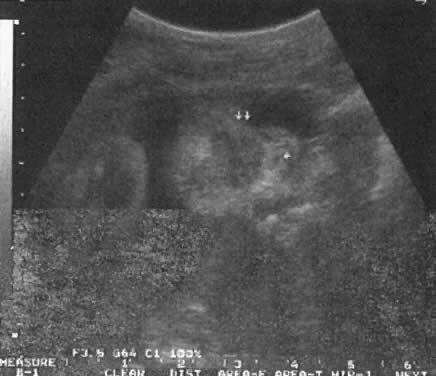
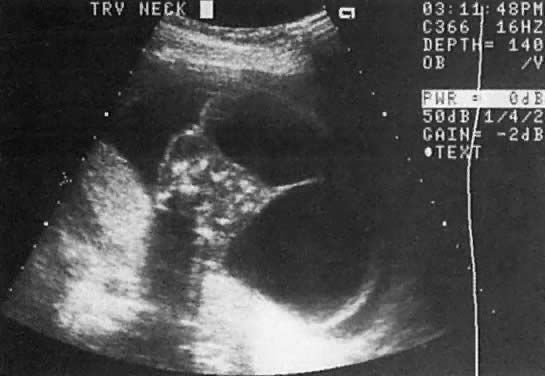
TIFFA examinations entail ultrasonic visualization of at least 34 fetal views and measurement of seven specific sites (Fig. 1). For each of these views, the ultrasonographer should indicate whether the structure is normal, previously normal, not applicable, suspect, abnormal, or poorly visualized. Poor visualization occurs when (1) fetal movement or position does not allow adequate imaging of a specific area, or (2) gestational age is less than 19 to 20 weeks.
|
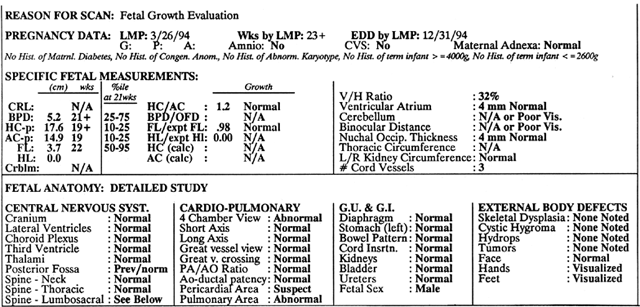
A normal TIFFA scan takes approximately 1 hour to complete. However, the presence of an anomaly is likely to extend the study and make it necessary for the mother to undergo a combination of genetic counseling, amniocentesis, or chorionic villi sampling to define fetal karyotype.
Studies that definitively resolve any possible concern regarding the long-term safety of ultrasound as well as its cost:benefit analysis are not yet available.6
Ewigman and co-workers7 evaluated the efficacy of routine antenatal diagnostic imaging with ultrasound (RADIUS) in reducing adverse perinatal outcome. Secondary outcomes included maternal morbidity and diagnostic accuracy of ultrasound. Study subjects were recruited from 92 obstetric and 17 family medicine practices in six states. Selection was based on predefined entry criteria that placed all study patients (screened group) in a low-risk category. Ultrasound screening examinations were performed on 7812 study subjects during two pregnancy intervals: 15 to 22 weeks, and 31 to 35 weeks. The women (n = 7718) in the control group had an ultrasound examination performed only if it was clinically indicated. The sensitivity in the detection of congenital anomalies was significantly higher in the screened versus control groups (34.8% vs 11%, p < 0.01); however, the higher detection of anomalies did not result in improved perinatal outcome. Romero8 and others9, 10 attribute the lack of improvement in perinatal outcome to (1) the limited sensitivity in the detection of anomalies in the RADIUS study, and (2) the low rate of pregnancy termination in women diagnosed with a fetal anomaly.
The low sensitivity (16.6%) in the detection of anomalies in the RADIUS study, particularly before 24 weeks' gestation, is very disturbing because it does not represent the current diagnostic capability of ultrasound. Specifically, in four studies11, 12, 13, 14 published from 1990 to 1992, the sensitivity of ultrasound screening for congenital anomalies before 24 weeks' gestation was 40%, 60.7%, 74.4%, and 84.3%, respectively. The specificity and negative predictive values were greater than 98%.11,12, 13, 14 Except for the Helsinki trial,11 the positive predictive values in these studies were equal to or greater than 98%.12, 13, 14