Cytogenetic studies can be performed readily from amniotic fluid cells, chorionic villus cells, or fetal lymphocytes. Thus, virtually all chromosomal disorders are potentially detectable in utero. Although technically feasible, however, it is not appropriate to determine the complement of every fetus because for many couples, the risks of prenatal diagnosis outweigh the potential benefits. Amniocentesis is considered to increase the risk of spontaneous abortion by approximately .5% over the background (discussed elsewhere in this volume.) CVS increases the pregnancy loss rate by approximately 1%; the risk associated with umbilical cord blood sampling probably is similar.11,12 In this section, unequivocal indications for cytogenetic studies are considered.
Advanced Maternal Age
One of the most common indications for antenatal cytogenetic studies is increased maternal age.32 Although there still is no unequivocal explanation for the relationship between aneuploidy and advanced maternal age, one factor could be that with advancing maternal age, there is decreasing maternal selection against chromosomally abnormal conceptuses.33 Given that approximately 7% of conceptuses but only .5% of liveborn infants are chromosomally abnormal, it is not unreasonable to suggest that maternal selection may be a factor in elimination of abnormal embryos. Another, more widely accepted, hypothesis is that chiasmata between homologous chromosomes decrease in aging oocytes, leading to nondisjunction and chromosomally abnormal ova. A more recent theory is that it is the decline in the oocyte pool, or in the number of maturing oocytes per cycle, that accounts for the increase in trisomies with advancing maternal age.34,35 In any case, in contrast to the overall incidence of trisomy 21 (1:800 live births in the United States),36 the likelihood of a 35-year-old mother having a child with trisomy 21 is 1:385; at 39 years of age, the risk is 1:137, and at 45 years of age, the risk is 1:30 (Table 2).37
Table 2. Risk of Having a Liveborn Child With Down Syndrome or Other Chromosomal
Abnormality
| Maternal Age (y) | Risk of Down Syndrome | Total Risk for All Chromosomal Abnormalities |
| 20 | 1/1667 | 1/526 |
| 21 | 1/1667 | 1/526 |
| 22 | 1/1429 | 1/500 |
| 23 | 1/1429 | 1/500 |
| 24 | 1/1250 | 1/476 |
| 25 | 1/1250 | 1/476 |
| 26 | 1/1176 | 1/476 |
| 27 | 1/1111 | 1/455 |
| 28 | 1/1053 | 1/435 |
| 29 | 1/1000 | 1/417 |
| 30 | 1/9520 | 1/384 |
| 31 | 1/9090 | 1/384 |
| 32 | 1/7690 | 1/322 |
| 33 | 1/6250 | 1/317 |
| 34 | 1/5000 | 1/260 |
| 35 | 1/3850 | 1/204 |
| 36 | 1/2940 | 1/164 |
| 37 | 1/2270 | 1/130 |
| 38 | 1/1750 | 1/103 |
| 39 | 1/1370 | 1/820 |
| 40 | 1/1060 | 1/650 |
| 41 | 1/8200 | 1/510 |
| 42 | 1/6400 | 1/400 |
| 43 | 1/5000 | 1/320 |
| 44 | 1/3800 | 1/250 |
| 45 | 1/3000 | 1/200 |
| 46 | 1/2300 | 1/150 |
| 47 | 1/1800 | 1/120 |
| 48 | 1/1400 | 1/100 |
| 49 | 1/1100 | 1/700 |
Because sample size for some intervals is relatively small, 95% confidence limits are sometimes relatively large. Nonetheless, these figures are suitable for genetic counseling.
*47,XXX excluded for ages 20–32 (data not available). (Data from Hook EB: Rates of chromosome abnormalities at different maternal ages. Obstet Gynecol 58:282, 1981; and Hook EB, Cross PK, Schreinemachers DM: Chromosomal abnormality rates at amniocentesis and in live-born infants. JAMA 249:2034, 1983)
Trisomy 21 is not the only chromosomal abnormality that increases with maternal age. Trisomy 13, trisomy 18, 47,XXX, and 47,XXY also show an increased mean maternal age.28 Based on these data, most United States authorities believe that prenatal diagnosis should be offered to all women who will be 35 years of age or older when their infant is born. However, the choice of a particular age is largely arbitrary because the risk for a chromosomally abnormal child increases steadily year to year, even among younger women. Therefore, flexibility is desirable when confronted by an inquiry from a woman younger than 35 years of age. Some women younger that 35 years of age may be relatively less concerned about the risk of abortion than the risk of a chromosomally abnormal liveborn infant and may wish to have a diagnostic procedure, despite the ostensibly unfavorable risk-to-benefit ratio.
Finally, it is worth emphasizing that the aforementioned risk figures are based on detection in liveborn infants. In fact, the incidence of abnormalities in antenatal studies at 16 to 18 weeks' gestation is approximately 50% higher than that in liveborn infants,37,38 and the incidence in first-trimester CVS studies is even greater.39 The discrepancies between the frequencies in liveborn infants and in first-trimester and second-trimester fetuses are accounted for by the disproportionate number of chromosomally abnormal fetuses that abort spontaneously before live birth.37,38,39,40
Maternal Serum Screening
Maternal serum alpha-fetoprotein (MSAFP) screening initially was developed for detection of fetal neural tube defects, which are associated with elevated values of MSAFP. However, in 1984, Merkatz and associates observed that fetal autosomal trisomy was associated with low MSAFP values.41 Why MSAFP is decreased in such pregnancies still is uncertain but probably relates to decreased fetal production of alpha-fetoprotein. Complicating screening, however, is that the median MSAFP level is decreased only slightly in pregnant women carrying fetuses with Down syndrome. Fortunately, other fetal–placental products have also proved useful for prenatal screening for Down syndrome in both the first and second trimesters (see also chapter 114). Initially studied in second trimester pregnancies, Bogart and colleagues showed that maternal serum human chorionic gonadotropin levels were significantly higher in pregnancies complicated by chromosome abnormalities.42 In fact, human chorionic gonadotropin was superior to MSAFP as a screening tool for chromosomal abnormalities. Canick and colleagues reported decreased levels of maternal serum unconjugated estriol in the second trimester in women with Down syndrome pregnancies.43 The authors suggested that decreased unconjugated estriol was related to immaturity of the fetal adrenal cortex, fetal liver, and placenta.
This suggested that screening would be most efficient if based on the aggregated values of maternal age, MSAFP, unconjugated estriol, and human chorionic gonadotropin (hCG). Using this combination of maternal serum markers and maternal age, Wald and colleagues retrospectively identified 60% of 77 Down syndrome pregnancies with a projected amniocentesis rate of 5%.44 In the first prospective trial of this approach in the United States, Haddow and associates reported screening more than 25,000 women.45 Patient-specific risk estimates were based on maternal age and serum screening, and patients were offered amniocentesis when the calculated risk for fetal Down syndrome was greater than or equal to 1 in 190 and gestational age was verified (3.8% of the population). Two thirds of known cases of Down syndrome were detected. Assuming that the prevalence of Down syndrome in the second trimester actually was higher but that some undetected abnormal fetuses were aborted spontaneously before live birth, the authors calculated a detection rate for Down syndrome of 58%. In a similar fashion, Phillips and colleagues have reported a prospective trial restricted to women younger than 35 years old.46 In this study, the risk cut-off was 1 in 274, with identification of 57% (4 of 7) of known cases of Down syndrome and an amniocentesis rate of 3.2%. A third prospective trial by Burton and colleagues used a cut-off of 1 in 270.47 With a 10.4% initial positive rate and an offered amniocentesis rate of 5.9%, 10 of 12 known cases of Down syndrome were identified. Increased risk for trisomy 18 also was identified with a different combination of cut-off values for the three serum markers (decreased MSAFP, unconjugated estriol and hCG). One abnormality was detected for every 33 amniocenteses performed, including two cases of trisomy 18 and one case of triploidy. In addition, three of the five cases of 45,X in the screened population were detected.
More recently, addition of inhibin A to the Down syndrome serum screening panel has been shown to increase detection efficiency.48 Further, urinary analytes, e.g., hyperglycosylated hCG, are being examined for usefulness in Down syndrome screening.49 In the first trimester, median free beta-hCG is elevated and pregnancy-associated plasma protein A (PAPP-A) is decreased in women with Down syndrome fetuses.50 These markers, combined with measurement of fetal nuchal translucency, are being studied in Great Britain and the United States to determine if first trimester screening is superior to screening in the second trimester.51,52
Second trimester serum screening has proved very useful for providing an individual, patient-specific risk for women younger than 35 years of age who typically would not be offered prenatal diagnosis. Whether maternal serum screening should be substituted for amniocentesis or CVS for women 35 years of age and older is a controversial issue.53,54,55 If invasive testing is offered only to women with abnormal serum screens rather than to all women older than 34 years of age, the detection rate of fetal Down syndrome is lower (89%), but only 25% of this population requires amniocentesis. Conversely, if all older women are offered and accept amniocentesis or CVS, the detection rate is 100%. Additionally, multiple marker screening is not highly sensitive in detecting chromosome abnormalities other than trisomy 21 and trisomy 18, which also increase with advancing maternal age. Thus, a significant number of aneuploidies are missed when using serum screening alone rather than invasive testing. Awaiting the results of second trimester multiple marker screening before offering invasive testing also frequently results in delay in the diagnosis of chromosome abnormalities until approximately 20 weeks' gestation, rather than detection in the late first or early second trimester after CVS or amniocentesis. Finally, justly or not, obstetricians are concerned about their legal liability should an older woman have a child with Down syndrome after a normal serum screen. For these reasons, the advantages and limitations of using serum screening as an alternative to invasive testing for cytogenetic abnormalities should be discussed carefully with patients before a choice is made.
Previous Child With Chromosomal Abnormality
After the birth of one child with either an autosomal trisomy or a sex chromosome abnormality, the likelihood that subsequent progeny will have a chromosomal abnormality traditionally has been considered increased, even if parental chromosome complements are normal. However, the risk for a second offspring with Down syndrome or another chromosomal abnormality appears to be increased only for mothers 29 years of age or younger at the time of the birth of the proband with Down syndrome (Table 3).56 Nonetheless, parental anxiety dictates that antenatal chromosomal studies at least be offered to all couples who have previously had a child with Down syndrome.
Table 3. Relationship of Recurrence Risk to Maternal Age at Birth of Proband
With Down Syndrome
| Maternal Age (y) at Birth of Proband | No. of Expected Cases Based on Maternal Age | No. of Observed Cases | Observed vs Expected p Value |
| ≤24 | 1.19 | 8 | 0.0004 |
| 25–29 | 1.59 | 6 | 0.0060 |
| 30–34 | 1.26 | 2 | 0.36*0 |
| 35–39 | 2 | 4 | 0.16*0 |
| ≥40 | 0.75 | 0 |
*Not significant. (Data from Mikkelsen M: Down syndrome: Current stage of cytogenic epidemiology. In Bonne-Tamir B, Cohen T, Goodman RM, (eds): Human Genetics, Part B: Medical Aspects, pp 297–309. New York, Alan R. Liss, 1982)
Information concerning recurrence risk after the birth of a child with a chromosomal abnormality other than trisomy 21 is very limited, but data from five collaborative studies indicate that the risk is 1% to 2% for either the same or a different chromosomal abnormality (Table 4).57,58,59,60,61 Thus, antenatal studies also should be offered to such couples.
| Proband | No. of Abnormals/Total (at Amniocentesis or CVS) | |
| Trisomy 13 | 4/596 | 2: +18; 1:+21; 1: t(Y;22) |
| Trisomy 18 | 20/1132 | 7: +21; 6: +13; 3:+18; 1: +9; 1: +12; 1:+15; 1: inv[18] |
| Other autosomal abnormalities | 4/256 | 2:XXY; 1: +21; 1: mos t[B;G] |
| Sex chromosomal abnormalities | 3/142 | 1: 45,X; 1: XYY; 1: +13 |
| Total | 2126 | 31(1.5%) |
CVS, chorionic villus sampling
(Data from Mikkelsen M; Stene J: Previous child with Down syndrome and other chromosome aberrations. In Murken JD, Stengel-Rutdowski S, Schwinger E, [eds]: Prenatal Diagnosis: Proceedings of the Third European Conference on Prenatal Diagnosis of Genetic Disorders, pp 22–23. Stuttgart, F Enke, 1979; Simoni G, Fraccaro M, Arslanian A, et al: Cytogenetic findings in 4952 prenatal diagnoses: An Italian collaborative study. Hum Genet 60:63, 1982; Stene J, Stene E, Mikkelsen M: Risk for chromosome abnormality at amniocentesis following a child with a noninherited chromosome aberration. Prenat Diagn 4 [special issue]: 81, 1984; Mikkelsen M: In Jackson L [ed]: CVS Newsletter, No. 19, pp 7–10. December 1, 1986; Jewell AF, Keene WE, Ferre MM, et al: Analysis of the recurrence risks for trisomy 13 and 18. Am J Hum Genet 59:A121, 1996
Parental Translocation, Inversion, or Aneuploidy
TRANSLOCATION
A third, less common, cytogenetic indication for antenatal diagnosis is the presence of a balanced translocation in a parent. The rare detection of an inversion or a numerical chromosomal abnormality (aneuploidy) warrants similar attention. The significance of a translocation can be illustrated by considering the most common type of translocation Down syndrome, a Robertsonian translocation between chromosomes 14 and 21 (Robertsonian translocations involve the acrocentric chromosomes: 13, 14, 15, 21, 22). If a child has Down syndrome resulting from such a translocation (e.g., 46,XX-14, + t[14q;21q]), the rearrangement originates de novo in 50% to 75% of cases (e.g., it is present in neither parent). The likelihood of Down syndrome recurring in the progeny of parents whose previous offspring had a de novo translocation probably is minimal, although recurrence of apparently de novo translocations (21q;21q) has been reported.62 Conversely, in 25% to 50% of subjects who have Down syndrome as a result of a translocation, one parent has the same translocation chromosome in a balanced state (e.g., 45,XX-14,−21, + t[14q;21q]). The theoretical risk that a parent carrying a t(14q;21q) chromosome will have a child with Down syndrome is 33%. However, empirical risks are considerably less. If the father carries the translocation, the risk is approximately 3%, whereas if the mother carries the translocation, the risk is approximately 10% to 15%. This sex-specific difference has been found in cases ascertained through chromosomally abnormal liveborn infants,63 as well as in collaborative reports of amniotic fluid studies64 and CVS60 (Table 5). Risks are considered similar for other Robertsonian translocations involving chromosome 21 (e.g., t[13q;21q], t[15q;21q], t[21q;22q]), but Robertsonian translocations that do not include chromosome 21 apparently carry much lower risks for unbalanced offspring. In fact, t(13q;14q), the most common Robertsonian translocation found in normal persons, apparently confers 1% to 2% risk (see Table 5).64 Liveborn offspring of individuals with balanced homologous translocations (e.g., 21q;21q or 13q;13q) will virtually all be trisomic for the involved chromosome.
| Fetus | ||||
| Rearrangement | Sex of Carrier | Normal | Carrier | Unbalanced |
| t(14q; 21q)* | Female | 035 | 055 | 14 (13.5%) |
| Male | 017 | 012 | 1 (3.3%)- | |
| t(13q; 14q) | Female | 039 | 096 | 1 (0.7%)- |
| Male | 012 | 054 | 2 (2.9%)- | |
| Reciprocal translocations (pooled) | Female | 235 | 365 | 76 (11.2%) |
| Male | 169 | 265 | 47 (9.8%)- | |
| Inversions (pooled) | Female | 009 | 079 | 3 (3.3%)- |
| Male | 008 | 074 | 0 (0.0%)- | |
(Data from Mikkelsen M: In Jackson L (ed): CVS Newsletter, No. 19, pp 7–10. December 1, 1986 and Daniel A, Hook EB, Wulf G: Risks of unbalanced pregnancy at amniocentesis to carriers of chromosomal rearrangements: Data from United States and Canadian laboratories. Am J Med Genet 31:14, 1989)
Reciprocal translocations do not involve centromeric fusion and, hence, usually do not involve acrocentric chromosomes. Unfortunately, because of their individual rarity, specific empirical data for most translocations are not available, and generalizations must be made on the basis of pooled data derived from many different translocations. Knowledge of the length of the translocated segment provides some additional guidance in predicting risk of a fetus with an unbalanced translocation, in other words, a longer translocation segment is associated with a lower risk.64 However overall, theoretical risks for abnormal (unbalanced) offspring are greater than empirical risks, which are approximately 10% for either maternal or paternal carriers (see Table 5).64
INVERSIONS
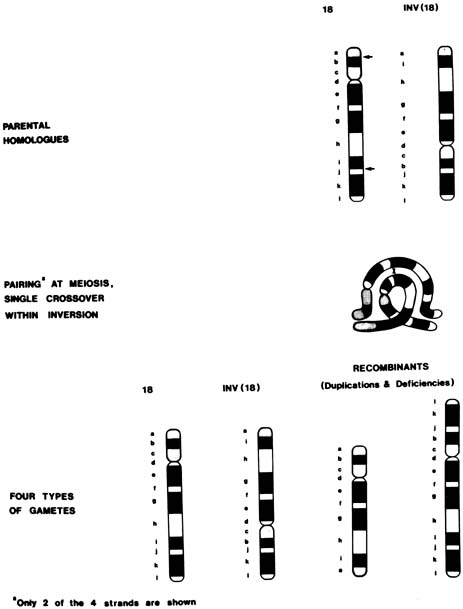
In a chromosomal inversion, the normal sequence of genes on the chromosome is altered. Subjects with such inversions are phenotypically normal; however, they may produce unbalanced gametes if, during meiosis I, crossing over (recombination) occurs within the inverted sequence. Thus, certain genes would be duplicated and others would be deficient in the unbalanced gamete (see Fig. 1). Pericentric inversions and inversions involving long segments are more likely to be associated with anomalous offspring than are paracentric or short inversion segments.65 Empirical data are not available for specific inversions, but pooled data for all inversions indicate approximately a 3% risk for abnormal progeny, with maternal carriers again at greater risk than paternal carriers39 (see Table 5). An exception is inv(9), which is a common variant and is thought to be without clinical significance.
|
ANEUPLOIDY
If a parent has a numerical chromosomal abnormality (aneuploidy), the risk to offspring is increased. For example, approximately 35% (but not 50%) of offspring of females with 47,XX, + 21 (Down syndrome) are aneuploid;66 therefore, antenatal chromosomal studies are indicated in a pregnant female with Down syndrome. Males with Down syndrome are sterile. If a parent is mosaic for trisomy 21, antenatal diagnosis is again in order.67,68 Although risk figures are plainly biased by the method of ascertainment, approximately 20% of offspring of fertile 45,X; 45,X/46,XX; and 45,X/46,XX/47,XXX subjects are said to show abnormalities.41 Women with 47,XXX or 46,XX/47,XXX also have produced children with chromosomal abnormalities, although most of the offspring are normal. Theoretically, 47,XYY men are also at increased risk for chromosomally abnormal offspring, and several abnormal offspring have been reported. Men with 47,XXY (Klinefelter syndrome) are sterile, but those with mosaicism (46,XY/47,XXY) may be fertile. Antenatal diagnosis should be offered to all aneuploid parents.
RELATIONSHIP OF ASCERTAINMENT TO EMPIRICAL RISK
Mode of ascertainment is a significant determinant of empirical risk for an unbalanced liveborn infant. Thus, when a family with a translocation is ascertained through a balanced proband, the risk for an unbalanced liveborn infant is very low. In contrast, if ascertainment is through an unbalanced individual, the risk for unbalanced offspring is significant.69,70 Presumably, then, with some translocations, unbalanced gametes do not arise during meiosis, or, alternatively, unbalanced products are selected against at the gametic or embryonic level. If a rearrangement has been ascertained during an evaluation for repetitive abortions, the risk for an unbalanced liveborn infant is lower than the risk expected after ascertainment through an anomalous liveborn infant but still is substantial.70
Fetuses Manifesting Intrauterine Growth Retardation or Anomalies on Ultrasound Examination
The potential indications considered previously are based on the premise that abnormal fetal outcome can be predicted on the basis of certain parental characteristics. However, trisomic fetuses, especially fetuses with trisomy 13 or 18, often show intrauterine growth retardation, which may be clinically evident during the second trimester. Clinical suspicion of intrauterine growth retardation can be followed with ultrasound monitoring to confirm intrauterine growth retardation. Gross anomalies also frequently can be visualized in fetuses with chromosomal abnormalities (Table 6).71–91 Antenatal chromosomal studies are appropriate if an abnormal fetus is detected on ultrasound examination. In addition to routine cytogenetic studies, particular defects suggest the need for more specific studies. For example, conotruncal heart defects are frequently associated with deletion of a small portion of chromosome 22 (del22q.11.2). FISH with specific probes will be diagnostic of this microdeletion that also implies the presence of other defects, for example, absent thymus and parathyroids (DiGeorge/velocardiofacial syndrome).92,93 Even if chromosomal studies cannot be obtained sufficiently early to permit pregnancy termination or termination is not desired, cesarean section for fetal distress in a fetus with a lethal abnormality might be avoided.
Table 6. Chromosomal Abnormalities in Pregnancies With Anomalies Detected
by Ultrasonography
| Ultrasound Finding* | No. of Abnormal/Total (% Abnormal) | Chromosome Results |
| Diaphragmatic hernia | 20/173 (11.6) | 12: + 18; 2: +21; 1: +21 +21; 1: triploid; 4: unbalanced autosomes |
| Duodenal atresia | 35/118 (29.6) | 30: + 21; 1: + 18; 1: + 13; 1: triploid; 1: unbalanced autosome; 1: XXX |
| Gastroschisis | 5/84 (6.0) | 2:+18; 1: + 13; 1: 45,X; 1: unbalanced autosome |
| Omphalocele | 78/319 (24.5) | 64: + 18; 6: +13 1: XXY; 1: triploid; 1: 45,X; 1: not specified; 4: unbalanced autosomes |
| Genitourinary | 43/443 (9.7) | 10: + 18; 8: +13; 7: + 21; 2: triploid; 1:45, X; 1 +9; 1: +8; 1: XYY; 8: unbalanced autosomes; 4: not specified |
| Cardiac | 129/473 (27.3) | 39: + 18; 25: + 21; 11: 45,X; 10: + 13; 1: + 9; 1: + 17; 1: triploid; 2: XXY; 4: unbalanced autosomes; 35: unspecified trisomy |
| Hydrocephalus | 55/402 (13.7) | 10: +21;10: + 18, 6: triploid, 6: + 13; 2: 45,X 1:XXX; 1:XXY: 16: unbalanced autosomes; 3: not specified |
| Choroid plexus cyst with no other anomalies seen | 28/723 (3.9) | 14: +18; 4: +21; 1:XXX; 1:XXY; 3:mosaic 45,X; 5: unbalanced autosomes |
| Holoprosencephaly | 21/44 (47.7) | 15: + 13; 1: + 18; 1: triploid; 4: unbalanced autosomes |
| Growth retardation and/or oligohydramnios | 112/780 (14.4) | 30: + 18; 23: triploid; 12: + 21; 9: + 13; 1 45,X; 34: unbalanced autosomes; 3: not specified |
| Polyhydramnios | 50/718 (7.0) | 15: +21; 18: + 18; 4: + 13; 1: triploid; 2: 45,X 1:XXX; 1: 46,X,i(Xq); 5: unbalanced autosomes; 3: not specified |
| Cystic hygroma | 183/296 (61.8) | 131: 45,X; 24: + 21; 13: + 18; 4: unbalanced autosomes; 2: +13; 1: triploid; 2: XXY; 6: not specified |
| Nonimmune hydrops without cystic hygroma | 86/254 (33.9) | 17: + 21; 5: + 18; 45: 45,X; 3: polyploid; 2: + 13; 5: unbalanced autosomes; 9: not specified |
*Listed under primary defect indicated by the investigator. In some cases, multiple abnormalities were present.
Several authors have reported that certain biometric findings (e.g., short femur, short humerus, pyelectasis, thickened nuchal fold, nuchal translucency, echogenic bowel, absent nasal bone) are indicative of an increased risk of fetal Down syndrome.94,95,96,97 The positive predictive value of the ultrasound findings depends on the patient's a priori risk based on maternal age or biochemical serum screening. Different recommendations have been made as to how best to estimate the absolute risk of Down syndrome.98 Antenatal diagnosis should be offered when the risk estimate is greater than the procedure-associated risk of pregnancy loss.
Mendelian Disorders Associated With Chromosome Breakage
Several inherited disorders are characterized by chromosome breakage in vivo and in vitro. Persons with these disorders often show increased propensity for neoplasia, growth retardation, and various somatic anomalies. Bloom's syndrome, ataxia-telangiectasia, and Fanconi's anemia are examples of such disorders. In some of these disorders, when the precise molecular defect is not known in individual families, distinctive cytogenetic features may permit antenatal diagnosis. For example, Voss and colleagues diagnosed Fanconi's anemia in a second-trimester fetus on the basis of high frequencies of spontaneous and clastogen-induced chromosome breakage in amniotic fluid cells.99 Similar studies have been performed with chorionic villus tissue100 and fetal blood.101 Parallel cultures of cells from other family members are required to distinguish affected fetuses from heterozygotes.102
Knowledge that patients with ataxia-telangiectasia (A-T) have an increased rate of spontaneous chromosome breakage has historically facilitated the diagnosis in the second trimester.103,104 However, localization of the A-T gene now allows more reliable molecular genetic testing.105 In Bloom's syndrome, the rate of sister chromatid exchanges is increased in peripheral lymphocytes, fibroblasts, and bone marrow cells, making prenatal diagnosis feasible even in families in which the gene mutation is unidentified. All these disorders are inherited in autosomal recessive fashion; therefore, couples who have had an affected child have a 25% recurrence risk in each pregnancy. Antenatal diagnosis should be offered to such families.
Fragile X Syndrome and Other X-linked Recessive Disorders
The fragile X syndrome is an X-linked disorder characterized in males by moderate mental retardation, macroorchidism, and a long face with a prominent jaw. Approximately one third of female carriers (heterozygotes) are mildly retarded, and the others have a normal phenotype. This syndrome accounts for a significant proportion of cases of familial X-linked mental retardation. The gene responsible for the condition is linked to a fragile site on the long arm of the X chromosome, visible as a break, or gap, in the chromosome structure. The fragile site is seen only when cells are grown in special medium deficient in folic acid and thymidine or when an antimetabolite such as 5-fluorodeoxyuridine or methotrexate is added to the culture medium.
Prenatal diagnosis has been accomplished by visualization of the fragile site in amniotic fluid cells, chorionic villus tissue, and fetal blood.106,107,108,109,110 Unfortunately, both false-negative and false-positive results have occurred in amniotic fluid and CVS samples.108,109,110 Molecular (DNA) methods also have been used to diagnose fragile X syndrome and have proven the more reliable approach.111,112
In other X-linked recessive, or male-limited autosomal dominant traits, only males are affected. It is possible to distinguish affected from unaffected male fetuses in some but not all of the sex-limited disorders. In others, affected infants can be avoided consistently only by terminating all pregnancies in which the fetus is male. In these cases, antenatal chromosomal studies to determine fetal sex may be indicated.