This category includes tumors that are purely composed of epithelial (sex cord) elements, mesenchymal (stromal) elements, or both. The descriptive term sex cord-stromal tumors avoids any controversy about the origin of the neoplastic constituents and, at the same time, acknowledges the frequent presence of both sex cord (e.g., granulosa and Sertoli cells) and stromal derivatives (e.g., fibroblasts, theca cells, and Leydig cells) within these tumors.
Most sex cord-stromal tumors are readily identifiable as belonging in either the granulosa-stromal cell or the Sertoli-stromal cell category, but a minority have patterns or cell types that are intermediate morphologically between the two. These tumors are designated sex cord-stromal tumors, unclassified. One tumor in this intermediate category with distinctive pathologic features and clinical associations has been given a specific designation: sex cord tumor with anular tubules. A rare tumor contains significant amounts of easily recognizable testicular and ovarian elements, warranting the diagnosis of a mixed form of sex cord-stromal tumor, or gynandroblastoma. That tumor is exceedingly rare if one uses the stringent criteria for its diagnosis adopted by WHO. Including the nonfunctioning fibroma, a pure stromal neoplasm that accounts for more than half of the sex cord-stromal tumors, the latter are responsible for only approximately 6% of all ovarian neoplasms.
Granulosa-Stromal Cell Tumors
These tumors are composed of granulosa cells and stromal derivatives, that is, fibroblasts and theca cells or both, with each type of cell occurring singly or in various combinations. Granulosa cell tumors are defined as neoplasms containing more than a minor component of granulosa cells with or without accompanying theca cells or fibroblasts. The designation granulosa-theca cell tumor has often been used for those specimens containing both cell types but granulosa cell tumor is probably more appropriate because it is difficult to be certain whether the theca element is truly neoplastic or reflects a response of the ovarian stroma to the proliferation of the neoplastic granulosa cells in many cases. The second group of tumors within the granulosa-stromal cell category has been designated generically as “tumors in the thecoma-fibroma group” because of the morphologic overlap between the thecoma and the fibroma and the occasional difficulty in distinguishing them when the morphologic features are intermediate.
GRANULOSA CELL TUMOR.
The granulosa cell tumor is the most common form of ovarian neoplasm associated with overt endocrine manifestations, which are almost always estrogenic. The tumor occurs in all age groups but is most common between 50 and 55 years of age. Fewer than 5% of the patients are younger than the age of normal puberty, and approximately three quarters of these children exhibit isosexual pseudoprecocity.4 In adult patients, menstrual disturbances such as menometrorrhagia, oligomenorrhea, and amenorrhea, which may last for months to years, are commonly encountered. The typical clinical manifestation in the postmenopausal woman is uterine bleeding. The pathologic background for these menstrual disorders comprises a spectrum of endometrial changes, which range from cystic hyperplasia to atypical hyperplasia to invasive adenocarcinoma. Carcinoma develops in approximately 5% of all adult patients with granulosa cell tumors (or thecomas), is twice as frequent in postmenopausal as in premenopausal women with these neoplasms, and is of low grade and amenable to curative therapy in the majority of cases.5 Granulosa cell tumors may be associated with additional estrogenic manifestations, such as swelling and tenderness of the breasts and a feeling of rejuvenation in older women. A rare granulosa cell tumor is virilizing.6 In approximately 10% of cases, the presenting manifestations are because of rupture of the tumor with hemoperitoneum.
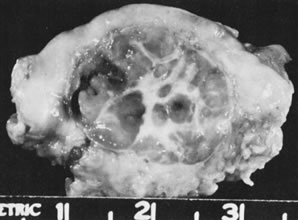
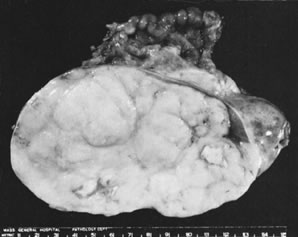
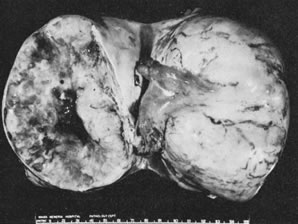
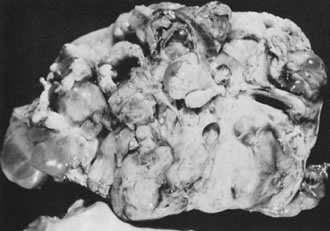
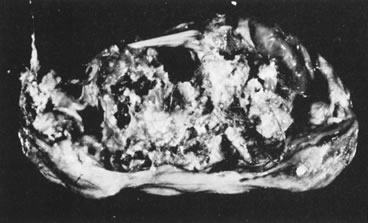
In 10% to 15% of the cases, the granulosa cell tumor does not cause palpable ovarian enlargement, and unless ultrasound examination is performed before surgery, is not detected until the time of a hysterectomy for another indication, such as atypical endometrial hyperplasia or carcinoma. More often, the tumor is palpable on pelvic or abdominal examination. At operation, it may appear solid or have both solid and cystic components (Fig. 1). On sectioning, the solid tissue may be gray, white, or yellow, reflecting the amount of intracellular fat it contains, and may be soft or firm depending on the relative quantities of granulosa cells and fibrothecomatous components. Two types of gross presentation are highly suggestive of granulosa cell tumor. One is a solid, pale yellow mass associated with extensive hemorrhage, and the other is a multicystic tumor with the locules filled with fluid or clotted blood. A rare appearance is that of a large unilocular or oligolocular thin-walled cystic tumor, resembling a serous cystadenoma; curiously, this gross appearance unusually often is associated with virilization instead of estrogenic changes.6 In summary, the granulosa cell tumor can simulate a variety of other types of ovarian tumor on gross examination, but the diagnosis may be suggested by the gross features, particularly in some clinical settings.
|
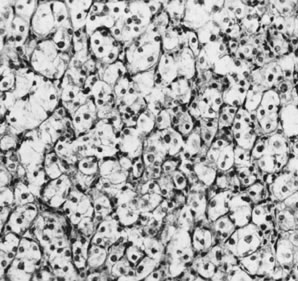
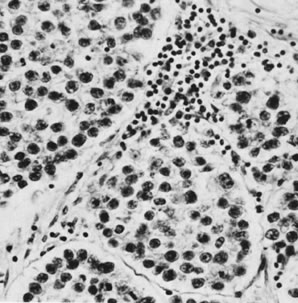
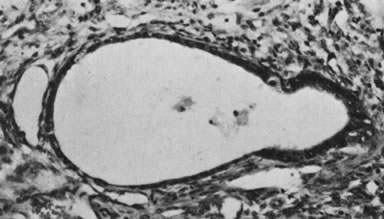
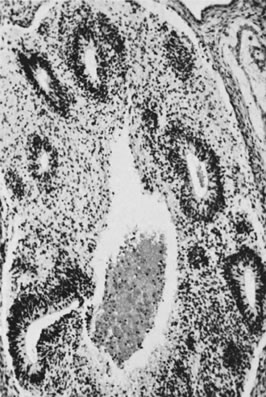
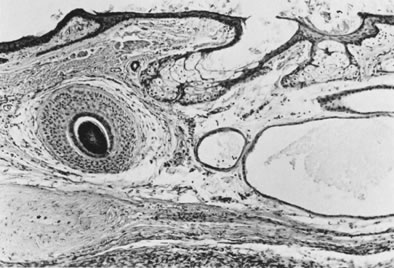
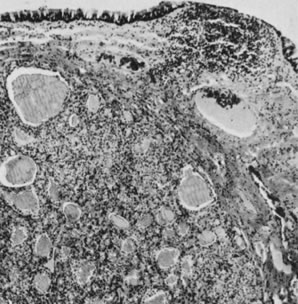
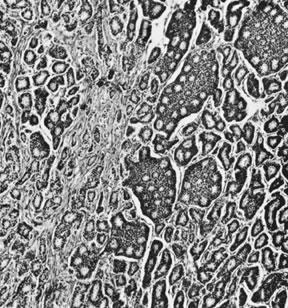
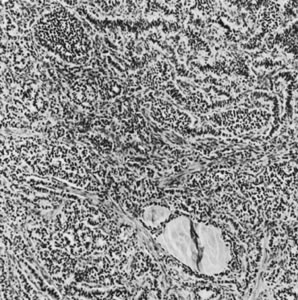
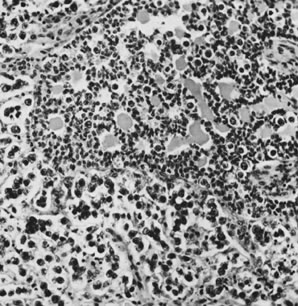
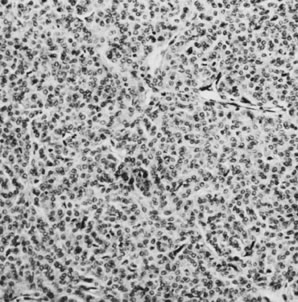
On microscopic examination, the granulosa cell tumor can be divided into two major subtypes, adult form and juvenile form, which differ strikingly in their patterns and cytologic features. The adult granulosa cell tumor is occasionally composed almost entirely of granulosa cells, with few or no accompanying thecal or fibroblastic elements, but more often the latter are present in some amount and may be substantial. The granulosa cell component of the tumor has a variety of patterns. The well-differentiated form most commonly has a microfollicular (Fig. 2), trabecular (Fig. 3), or insular pattern, or a combination of the three. Rarely, the pattern is macrofollicular, with the tumor composed of large follicles resembling normally developing follicles. Less well-differentiated tumors typically have a diffuse or sarcomatoid pattern, characterized by a sea of cells with little or no intervening stroma (Fig. 4); this type of tumor is particularly apt to rupture. Sometimes a watered-silk or zigzag (gyriform) pattern is seen. The stromal element of the tumor may consist of fibroblasts that have laid down considerable collagen, as well as cells that resemble theca externa, theca interna, or theca lutein cells.
|
|

|
Lipid stains and a variety of histochemical reactions, which, when positive, are considered characteristic of steroid-hormone-producing cells, typically color the theca cell component of the tumor but not the granulosa cells, suggesting that the theca cells are responsible for the hormone secretion. It is possible, however, that the theca cells secrete androgens, which are aromatized to estrogens by the granulosa cells in tumors containing both elements, as in the normal follicle. Some estrogenic granulosa cell tumors, in contrast, appear to have only a minor theca cell component, and the mechanism of estrogen production by such tumors is not clear.
The juvenile granulosa cell tumor is so named because more than 95% of the cases occur within the first 3 decades, accounting for the majority of granulosa cell tumors occurring during that age period.4 Microscopic examination generally shows a nodular pattern of granulosa cell proliferation with the nodules typically containing follicles intermediate in size between the microfollicles and macrofollicles of the adult form of the tumor. The follicles are typically more variable in size and shape than those of the adult granulosa cell tumor. The neoplastic granulosa cells typically have abundant cytoplasm (i.e., are luteinized) unlike the more commonly nonluteinized cells of the adult tumor. They usually exhibit brisk mitotic activity. In addition, the nuclei of the juvenile tumor are not pale and grooved as in the adult granulosa cell tumor and may be strikingly pleomorphic. As a result, some juvenile granulosa cell tumors have a highly malignant appearance microscopically and are sometimes confused with yolk sac tumors or embryonal carcinomas by inexperienced pathologists.
The treatment of granulosa cell tumors depends on their stage and the age of the patient.7 The majority of these tumors are stage I. Because bilaterality is encountered in fewer than 5% of the cases, unilateral tumors can be treated effectively by simple salpingo-oophorectomy when the patient is young and the preservation of fertility is an important consideration. For the older woman, the appropriate treatment is almost always hysterectomy with bilateral salpingo-oophorectomy, even in stage IA cases. Recurrences are mostly in the pelvis and lower abdomen, but distant metastases have been reported at numerous sites. Recurrences can often be treated effectively by another surgical procedure followed by chemotherapy or radiation therapy, to which some adult granulosa cell tumors have been very sensitive. Various chemotherapeutic regimens used in the treatment of malignant germ cell tumors, such as vincristine, actinomycin D, and cyclophosphamide; cisplatin, vinblastine, and bleomycin; and bleomycin, etoposide, and cisplatin, have proved effective in the treatment of extra-ovarian disease in cases of adult granulosa cell tumor. There has been too little experience with juvenile granulosa cell tumors to draw conclusions about the value of chemotherapy or radiation therapy.
The survival rate of patients with adult granulosa cell tumors is approximately 90% at 10 years but drops to approximately 50% at 30 years because of the frequency of late recurrences, which have been reported more than 30 years after surgery.8–10 Despite their more malignant histologic appearance, juvenile granulosa cell tumors are associated with a better prognosis, with a survival rate of approximately 90%, and a rarity of late recurrences.4 Recently, inhibin11 has become available to aid in detecting the presence of recurrent granulosa cell tumor, and immunostaining for inhibin may be of great value in the pathologic differential diagnosis of granulosa cell tumors and, indeed, other sex cord-stromal tumors.12
TUMORS IN THE THECOMA-FIBROMA GROUP.
This category includes fibromas and thecomas, as well as tumors with intermediate features. Included in the last category is a distinctive subtype: the sclerosing stromal tumor.
Fibroma.
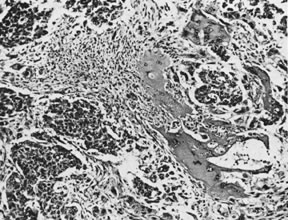
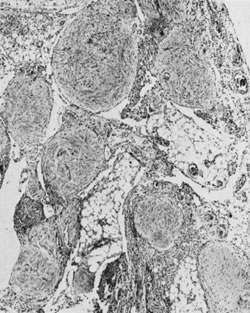
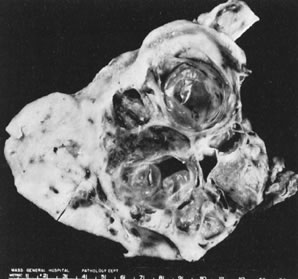
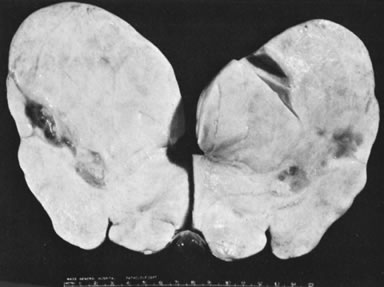
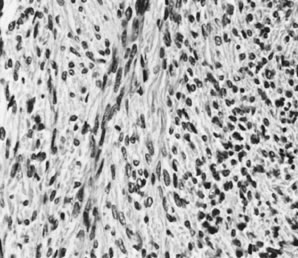
Ovarian fibromas occur at an average age of 48 years; fewer than 10% are encountered in females younger than the age of 30 years. These tumors are solid and have a fat, chalky-white, trabeculated appearance on their sectioned surfaces (Fig. 5). They are bilateral in approximately 10% of the cases.13 Microscopic examination shows intersecting fascicles of spindle cells producing collagen (Fig. 6); a storiform or pinwheel pattern is occasionally present. Hyaline plaques and foci of calcification are sometimes encountered, with calcification occurring, particularly in the bilateral tumors that are found in young women with the basal cell nevus (Gorlin's) syndrome.14 Intercellular edema is often present; a marked degree of edema has been correlated with the presence of ascites, which accompanies 40% of fibromas over 10 cm in diameter and with Meigs' syndrome (ascites and hydrothorax relieved by the removal of a benign ovarian tumor with fibromatous features), which occurs in fewer than 1% of the cases.15 Cellular fibromas with minor mitotic activity may recur, especially if they are found to be ruptured or adherent at the time of operation; even in the absence of rupture or adhesion, cellular fibromas occasionally recur many years after their removal.16 The rare fibrosarcoma is characterized by significant nuclear atypicality and mitotic activity that is generally greater on the average than three per 10 high-power fields; these tumors are highly malignant.
|
|
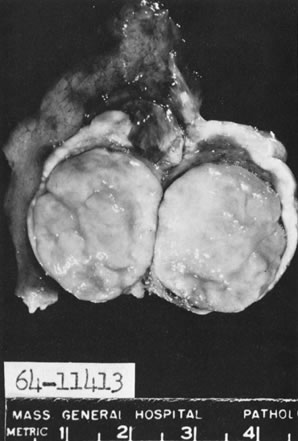
Thecoma.
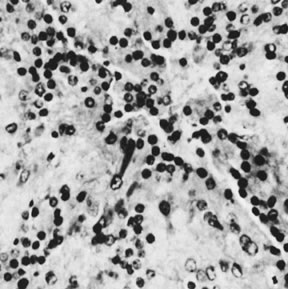
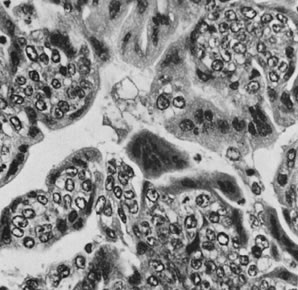
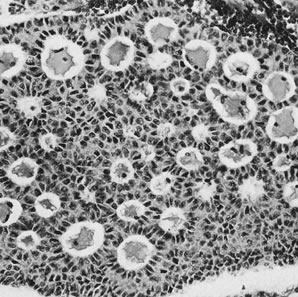
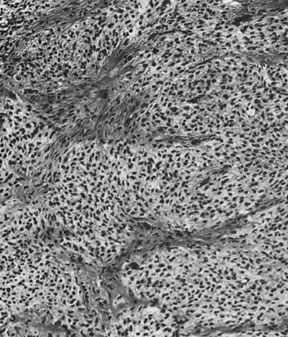
Thecomas are divided into two subtypes: the typical and the luteinized forms.17,18 Both tumors form solid masses composed of firm tissue that may be yellow throughout or white with focal areas of yellow coloration (Fig. 7). Microscopic examination of the typical thecoma shows the exclusive presence or marked predominance of pale, vacuolated, lipid-rich cells that resemble theca interna cells (Fig. 8). A fibromatous component may be present, and occasionally a minor number of granulosa cells are observed. The luteinized thecoma has a background of typical fibroma or thecoma but contains, in addition, large lutein cells (i.e., cells resembling those of the corpus luteum) (Fig. 9). The lutein cells may contain variable amounts of lipid in their cytoplasm.
|
|
|
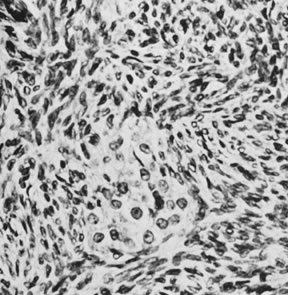
The typical thecoma occurs rarely before puberty and is seen in somewhat older patients on the average than the granulosa cell tumor. The clinical manifestations, which are almost always estrogenic, are similar to those of the granulosa cell tumor, except that a malignant behavior is extremely rare. Among the cases that have been described as malignant in the literature are endocrinologically inert fibrosarcomas and diffuse granulosa cell tumors misinterpreted as thecomas.19 Luteinized thecomas occur at an average younger age than typical thecomas; although they are most frequent in postmenopausal women, 30% of them have been encountered in patients younger than 30 years of age. The luteinized thecoma is estrogenic in approximately half the cases and androgenic in 11%. Because the thecoma is almost invariably benign, it can be treated by a conservative operation when it occurs in a young woman. A unique ovarian tumor that appears to fall in the luteinized thecoma group is associated with sclerosing peritonitis.20 The tumor is often bilateral and typically exhibits brisk mitotic activity. Despite the latter, it has not exhibited metastatic potential, but rare tumors have been fatal as a result of complications of the sclerosing peritonitis they cause by currently unknown mechanisms.
Unclassified.
Because the theca cell and fibroblast, both in the normal ovary and in ovarian tumors, are both derivatives of the ovarian stromal cell, it is not surprising that occasional tumors exist in an intermediate zone between the thecoma and the fibroma. Such tumors may contain an intermediate quantity of cytoplasmic lipid and be unaccompanied by clear-cut evidence of steroid hormone production. The designation of tumors in this category as thecomas by some observers and fibromas by others probably accounts for the considerable variation in the reported frequency of the thecoma relative to the granulosa cell tumor.
The sclerosing stromal tumor belongs in the intermediate category between the thecoma and fibroma but possesses distinctive clinical and pathologic features. 21 Unlike the fibroma and the typical thecoma, it has been encountered in the first 3 decades in more than 80% of personally observed cases. Ascites has complicated the sclerosing stromal tumor rarely, and exceptionally, estrogenic or androgenic manifestations have been described. The tumors have been unilateral. On gross examination, the neoplasm is typically predominantly solid with areas of edema and often cyst formation; an occasional tumor is predominantly cystic. The neoplastic tissue is white with foci of yellow coloration. The microscopic picture is characterized by the presence of pseudolobules of cellular tissue separated by cell-poor fibrous or edematous areas. The pseudolobules often contain a rich network of thin-walled vessels. The tumor cells are of two types: spindle cells producing collagen and rounded lutein-type cells with shrunken nuclei and vacuolated cytoplasm that contains considerable lipid. No case of malignant sclerosing stromal tumor has been reported. A rare cellular stromal tumor that contains signet-ring-like cells that do not contain mucin or lipid has been designated signet-ring stromal tumor.22
Sertoli-Stromal Cell Tumors (Androblastomas)
These tumors,23 which were originally designated arrhenoblastoma, have been divided into three categories according to their degrees of differentiation; a fourth category, characterized by the presence of foreign (heterologous) elements; and a fifth category with an architecture resembling that of the rete testis.
Sertoli-stromal cell tumors occur predominantly in young women in the reproductive age group, with an average age incidence of 25 years, but may be encountered occasionally in children and older women. Typically, these tumors are associated with a more-or-less abrupt onset of defeminization, with amenorrhea followed by progressive virilization, as a result of the production of a variety of androgens. A return of the menses to normal in approximately 1 month and usually some regression of hirsutism follows the complete removal of the tumor, but a decrease in size of an enlarged clitoris and restoration of normal female pitch occur less often. Although one third to two fifths of Sertoli-stromal cell tumors are virilizing, some are nonfunctioning and still others are associated with estrogenic manifestations. The latter may be the result of estrogen production by Sertoli cells or Leydig cells or possibly, in some cases, by secretion of androstenedione by Leydig cells and its conversion to estrone in the peripheral tissues.
The gross features of Sertoli-stromal cell tumors vary greatly from one specimen to another, resembling those of granulosa cell tumors, but the former tumors are cystic less often than the latter.
WELL-DIFFERENTIATED FORMS.
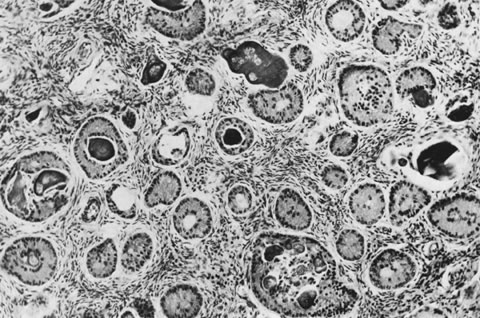
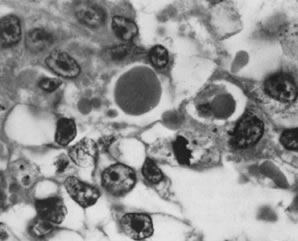
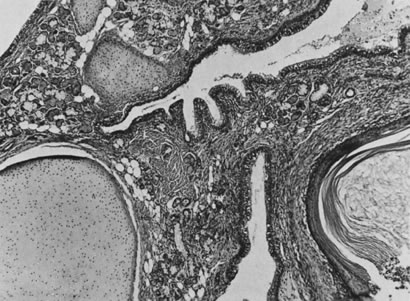
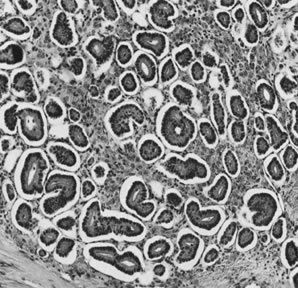
These tumors may be composed entirely or almost entirely of either Sertoli cells or Leydig cells or may contain both elements in considerable numbers; pure Sertoli cell tumors are rare.24 Well-differentiated Sertoli-stromal cell tumors of the mixed variety are characterized by hollow or solid tubules separated to varying extents by mature Leydig cells (Fig. 10), which occasionally contain rod-shaped eosinophilic bodies known as crystals of Reinke.25 These structures are more or less specific for Leydig cells. Varying amounts of lipid may be found within the cytoplasm of the Sertoli cells of the tubules as well as in the Leydig cells. These tumors may be associated with androgenic or estrogenic manifestations. Tumors composed mainly of Sertoli cells are often estrogenic. The pure Leydig cell tumor belongs nosologically in the Sertoli-stromal cell category but is discussed with steroid cell tumors because it is closest morphologically to neoplasms in that category.
|
TUMORS OF INTERMEDIATE DIFFERENTIATION.
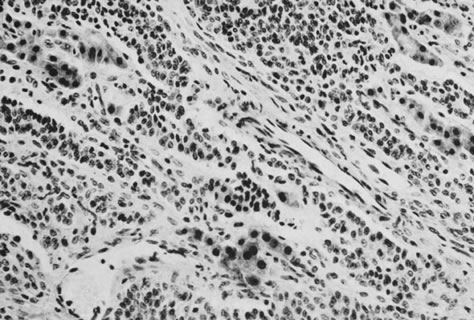
These tumors, which are the most common form of Sertoli-stromal cell tumor, are characterized by immature Sertoli cells, typically containing small oval-to-round nuclei and growing in a variety of patterns, and by a stromal component that varies in its cellularity, most typically being relatively acellular and often edematous, and contains at least some well-differentiated Leydig cells (Fig. 11). A lobular growth is often conspicuous on low power. The Sertoli cells may be disposed in round, oval, or elongated aggregates; in masses or within tubules, but most characteristically, they lie in short, single rows that simulate the sex cords of the developing testis. Varying amounts of lipid may be present in the neoplastic cells. The tumors are usually associated with androgenic manifestations but may be estrogenic or nonfunctioning.
|
POORLY DIFFERENTIATED TUMORS.
These tumors were originally called sarcomatoid because one of the most frequent forms contains areas resembling a fibrosarcoma. However, the Sertoli cell, as well as the stromal component of the tumor, may be poorly differentiated; and simulate an undifferentiated carcinoma, not otherwise specified;a poorly differentiated Sertoli cell tumor may be estrogenic. Minor better-differentiated areas may be crucial in categorizing these tumors.
TUMORS WITH HETEROLOGOUS ELEMENTS.
Heterologous elements are found mainly in tumors that have a background of intermediate or poor differentiation.26,27 The most commonly encountered component is mucinous epithelium resembling that of the gastrointestinal tract, containing goblet cells and even argentaffin cells. The mucinous component may be benign, borderline, or carcinomatous and rarely is the predominant element of the tumor on both gross and microscopic examinations. Occasionally, microcarcinoid tumors develop from the argentaffin cells. Islands of immature cartilage and rhabdomyoblasts are the mesenchymal heterologous element that may be encountered. They may be prominent components of the sarcomatoid form of the neoplasm.
RETIFORM SERTOLI-LEYDIG CELL TUMOR.
This tumor28 differs from the other forms clinically and pathologically, occurring at an average patient age of 15 years, 10 years younger than the average age of patients with Sertoli-stromal cell tumors as a group. Microscopic examination shows a network of tubules and papillae simulating the structure of the rete testis. Because of the presence of tubules, cysts, and papillae, the tumor is often misinterpreted by pathologists as a borderline or invasive serous tumor. The usual young age of the patient is a clinical clue that the tumor is not a serous tumor.
The prognosis of Sertoli-stromal cell tumors is related to both their stage and their histologic subtype.23 The well-differentiated tumors are benign. The rare tumors of other types that present at a stage higher than stage I are associated with a very poor prognosis. Patients with stage I tumors of intermediate differentiation have a survival rate of almost 90%; those with poorly differentiated tumors, 40%; and those with tumors containing mesenchymal heterologous elements such as skeletal muscle, cartilage, or both, approximately 30%. The presence of mucinous epithelium and carcinoid tumorlets does not appear to affect the prognosis of the underlying homologous component of the tumor. Too few cases of retiform Sertoli-stromal cell tumor with adequate follow-up for evaluation of prognosis have been reported, but these tumors appear to be more malignant clinically than otherwise similar Sertoli-stromal cell tumors without a retiform component.
Sertoli-stromal cell tumors are almost always unilateral, and therefore, unilateral salpingo-oophorectomy is the appropriate procedure for a stage IA tumor in a young patient in whom the preservation of fertility is an important consideration. The poor prognosis associated with poorly differentiated stage IA tumors, whether homologous or heterologous, raises the possibility of adjuvant chemotherapy to improve survival. In addition, because the survival of patients with ruptured intermediate tumors is approximately 20% lower than that of patients with intact tumors, adjuvant chemotherapy is a consideration for those tumors as well. There is very little experience with the chemotherapy of high-stage or recurrent Sertoli-stromal cell tumors, but partial and complete responses have been recorded occasionally with the use of regimens similar to those used for recurrent or metastatic granulosa cell tumors.
Gynandroblastomas
The term gynandroblastoma was coined to describe those tumors that contain cellular elements of both ovarian and testicular types.1 Unfortunately, the cases reported in the literature collectively comprise a hodgepodge of tumors that have little in common morphologically. In addition, it must be emphasized that extensive sampling of a granulosa cell tumor often discloses small areas that are more compatible morphologically with a Sertoli-stromal cell tumor and vice versa. For these reasons, the WHO has recommended that the term gynandroblastoma be restricted to those specimens in which there are substantial amounts of mature and indisputable ovarian and testicular cellular components. There are no consistent endocrine manifestations of these very rare tumors.
Sex Cord-Stromal Tumors, Unclassified
These tumors lie in the indeterminate zone between granulosa-stromal cell tumors and Sertoli-stromal cell tumors, with cells and patterns of growth that are compatible with, but not specific for, a diagnosis of either type of tumor. These neoplasms account for approximately 10% of all sex cord-stromal tumors. Sex cord-stromal tumors from pregnant patients are particularly apt to be hard to classify and often must be placed in the unclassified group.29
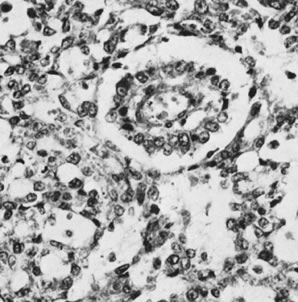
The sex cord tumor with annular tubules30 is a relatively rare neoplasm that exists in two forms: as a large solitary mass, which often metastasizes to lymph nodes, and, more remarkably, as a common feature of the Peutz-Jeghers syndrome (gastrointestinal polyposis with oral and cutaneous melanin pigmentation). In patients with this syndrome, the tumors are typically small, multiple, and focally calcified. The largest tumor associated with the Peutz-Jeghers syndrome was 3 cm in diameter. Microscopic examination shows solid tubules with a central mass of cytoplasm and peripheral nuclei; the tubules are arranged in the form of a simple ring with an eosinophilic hyaline body at the center or, more often, a complex ring with the tubule rotating around multiple hyaline spheres (Fig. 12). The small tumors in patients with the Peutz-Jeghers syndrome may be associated with estrogenic manifestations. The large tumors, which are not associated with the syndrome, are usually nonfunctioning but may have estrogenic or, rarely, androgenic manifestations and often secrete progesterone in large amounts, resulting in a decidual reaction in the endometrium. In addition to an elevation of progesterone in the serum, there may be an increase in müllerian inhibiting substance. In one reported case, it served as a helpful tumor marker, becoming elevated before clinical evidence of recurrent tumors over a period of 22 years.31
|