Ovarian malignancies in children may represent an array of unique problems for the clinician who is more accustomed to diagnosing and treating ovarian neoplasia in adults. Although ovarian malignancies in children are rare (representing only 0.2% of all ovarian neoplasms), their recognition and diagnosis are vital because they can be fulminant if treated inadequately. Advances in combination chemotherapy have enabled patients to survive the disease with tolerable (less toxic) short-term and long-term morbidity. Whenever possible, treatment should be individualized to preserve reproductive and menstrual function without jeopardizing the life of the child.
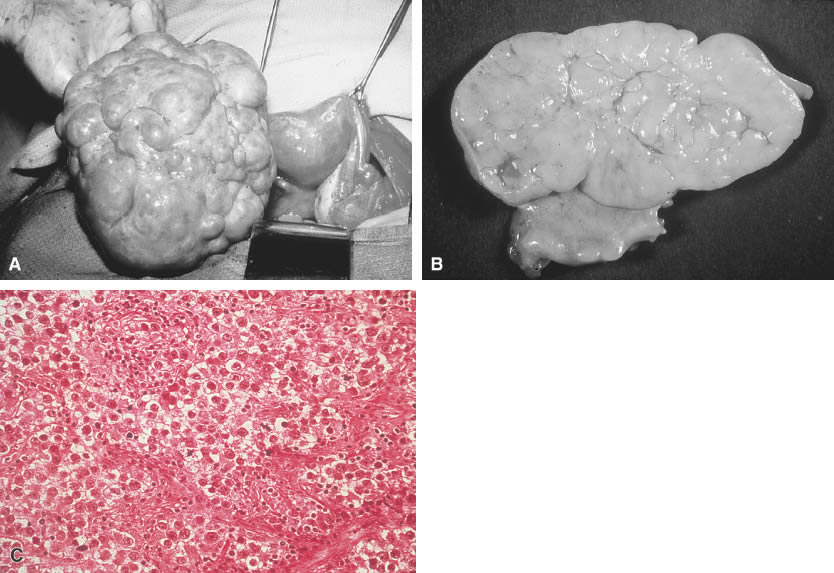
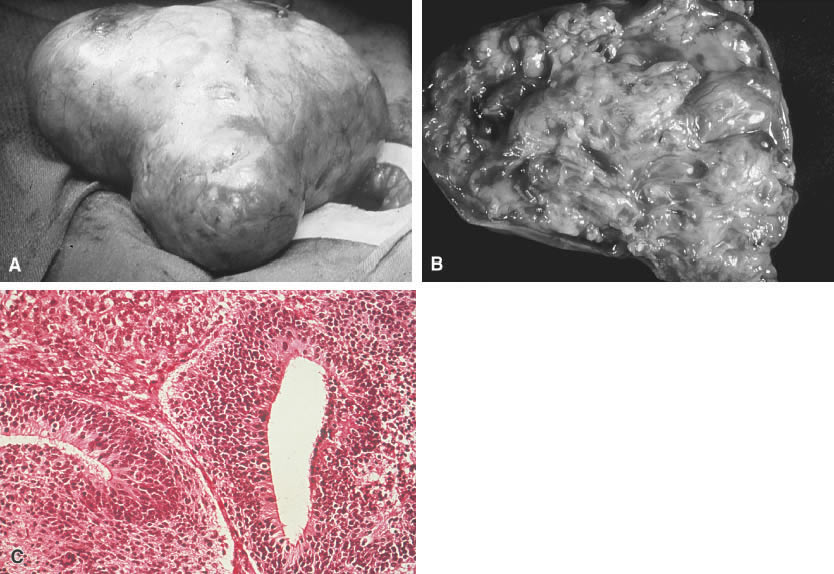
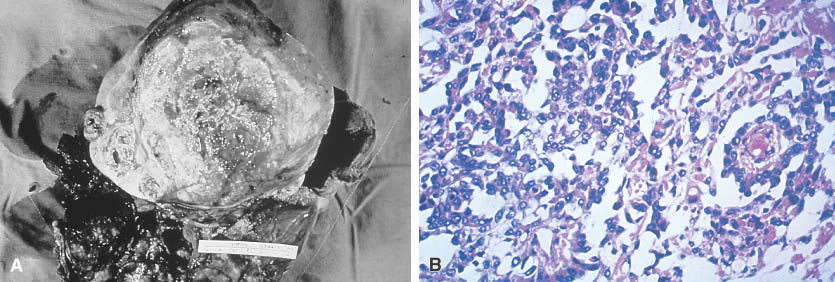
Tumors of germ cell origin (e.g., mature teratoma, malignant teratoma, endodermal sinus tumor [yolk sac tumor], embryonal carcinoma, dysgerminoma, primary choriocarcinoma) constitute approximately 70% of ovarian tumors in children (Table 1).1 All such tumors with the exception of the mature teratoma are malignant. The dysgerminoma in its pure form is considered a low-grade malignancy. Because these tumors have a common histogenesis, mixed germ cell tumors can be found, which yield a confusing clinical picture with regard to anticipated post-therapy responses.
TABLE 1. Incidence of Ovarian Malignancies in 874 Children According to
Histologic Tumor Types
Diagnosis | N | Malignant Tumors (%) |
Germ Cell |
||
Dysgerminoma |
214 |
24.5 |
Immature teratoma |
172 |
19.7 |
Mixed germ cell |
73 |
8.4 |
Endodermal sinus |
120 |
13.7 |
Embryonal |
25 |
2.9 |
Primary choriocarcinoma |
8 |
0.9 |
Total |
612 |
70.1 |
Sex-Cord Stromal |
||
Pure granulosa cell |
46 |
5.3 |
Juvenile granulosa cell |
30 |
3.4 |
Granulosa-theca cell |
21 |
2.4 |
Sertoli-Leydig cell |
23 |
2.6 |
Fibrosarcoma |
5 |
0.6 |
Sarcoma |
3 |
0.3 |
Pure Sertoli cell |
3 |
0.3 |
Nonspecific |
17 |
1.9 |
Total |
148 |
16.8 |
Surface Epithelial-Stromal Tumors |
||
Serous cystadenocarcinoma |
23 |
2.6 |
Mucinous cystadenocarcinoma |
20 |
2.3 |
Clear cell adenocarcinoma |
8 |
0.9 |
Endometrioid adenocarcinoma |
2 |
0.2 |
Adenocarcinoma |
13 |
1.5 |
Total |
66 |
7.5 |
Metastatic and Unclassified |
||
Lymphoma |
19 |
2.2 |
Adrenal rest |
1 |
0.1 |
Metastatic |
3 |
0.3 |
Unclassified |
25 |
2.9 |
Total |
48 |
5.5 |
TOTAL |
874 |
Surface epithelial stromal tumors (common epithelial tumors) are rare in children and represent the same morbidity and mortality in children as in adults; therapy also is similar to that used in adults. Granulosa-stromal cell tumors are the most common of the sex-cord stromal cell tumors, vary greatly in their clinical course, and are rare in children. This chapter reviews the world literature on the clinical characteristics and modern management of the aforementioned tumors, using the new histologic classification of ovarian tumors developed by the World Health Organization (Table 2).
TABLE 2. Histologic Classification of Ovarian Tumors*