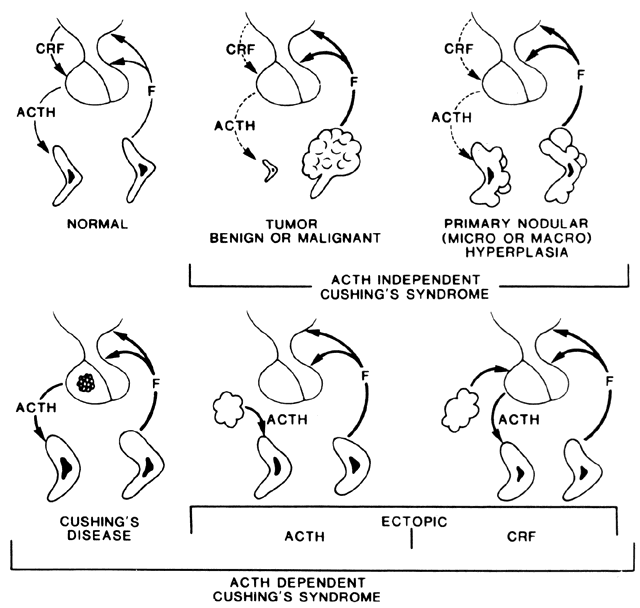
Congenital adrenal hyperplasia (CAH) is a group of common inborn errors of metabolism that are transmitted as autosomal-recessive traits.1,2 These enzymatic deficiencies cause impaired cortisol production as a result of their intermediary role in the steroidogenic pathway that converts cholesterol to cortisol. When cortisol production is decreased, the level of adrenocorticotropic hormone (ACTH) rises because of the absence of negative feedback.3 Because the body attempts to maintain normal cortisol levels, there is overproduction and accumulation of cortisol precursors proximal to the enzymatic block. These precursors are shunted toward the production of C19 androgens (androstenedione and testosterone), leading to the clinical manifestations of virilization and hirsutism. The 21-hydroxylase (21-OH) and 11β-hydroxylase enzymes also are used in the mineralocorticoid pathways whose end product is aldosterone. When the enzyme is defective in this pathway, patients cannot conserve sodium. This condition leads to the salt-wasting form of the disease.
Patients with the classic neonatal forms have sexual ambiguity, progressive virilization, and sometimes salt loss or hypertension. These conditions require life-long medical management4 and often surgical reconstruction of the deformed genitalia.5 The nonclassic (asymptomatic) and late-onset (usually pubertal) forms are more common than the classic presentation. The most common form of classic CAH is 21-hydroxylase deficiency (CAH21), which accounts for 95% of the cases of CAH. The next most common forms are 3β-hydroxysteroid dehydrogenase deficiency (CAH3β-HSD) and 11β-hydroxylase deficiency (CAH11β).2
21-Hydroxylase Deficiency
In CAH21, the abnormality is in the 21-OH enzyme (P45021), blocking the conversion of hydroxyprogesterone (17OHP) to 11-deoxycortisol (compound S). Basal circulating levels of both progesterone and 17OHP are elevated in the classic form, but usually only after ACTH stimulation in the cryptic, heterozygote, or attenuated (late-onset) varieties of CAH21.
GENETICS.
The different types of CAH21 can be attributed to multiple allelic variations encoding the cytochrome P450 enzyme, which performs 21-hydroxylation (CYP-21). Molecular studies showed two copies of the gene on the short arm of chromosome 6 in the class III region of the human lymphocyte antigens (HLA) complex (Fig. 1). The HLA genetic region encodes genes that have multiple allelic variants that determine histocompatibility. Other genes are located in the class III region, including the 21-hydroxylase genes, two complement factor 4 genes (C4), properidine factor B gene (Bf), and complement factor 2 gene (C2). The two 21-hydroxylase genes (CYP-21A and CYP-21B) alternate with the two genes that encode C4. The class III region of the HLA complex is located between the HLA class II region DR gene and the class I region tumor necrosis factor and HLA-B genes.6,7,8,9 The CYP-21B gene is functionally important, but the CYP-21A gene is a pseudogene; the gene product is enzymatically inactive.9,10,11
|
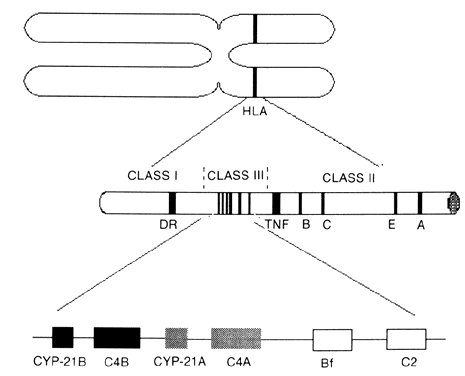
Gene deletions and substitutions in the CYP-21B gene affect P45021 function. Because the HLA genes are located in the same portion of chromosome 6 as the CYP-21 genes, certain allelic HLA variants are inherited together (genetically linked) with specific CYP-21 mutations. The HLA haplotype A1 B8 DR3 is associated with a CYP-21A gene deletion, but patients are phenotypically and hormonally normal. In contrast, the A3 BW47 DR7 haplotype, in which the CYP-21B gene is deleted,12 is associated with the salt-wasting form of CAH.13 Werkmeister and colleagues11 showed that 25% of patients with classic CAH carried alleles that had CYP-21B gene deletions, despite varied HLA compositions. The remaining 75% had smaller de novo point mutations that caused amino acid substitutions in the CYP-21B gene, thereby rendering the enzyme nonfunctional.
Seventy-four percent of the mutations are caused by gene conversions from CYP-21A to CYP-21B.14 Deletions or further duplications of the C4 and CYP-21 genes can occur as a result of unequal crossover due to misalignment of close spacing of homologous genes during meiosis.11,15 Triplication of the C4 and CYP-21 genes most frequently is associated with the late-onset form of CAH21.16 The B14 DR1 haplotype that is associated with 78% of cases of late-onset CAH21 features an extra C4B and CYP-21A gene as well as a valine-to-leucine mutation at codon 281 of the CYP-21B gene that normally is present in the CYP-21A gene. This codon in the CYP-21B gene is conserved in several mammalian species.14,17 The resultant amino acid change promotes a break in an α-helix that is required for optimal enzyme function. Other mutations transferred from the CYP-21A to the CYP-21B gene have been identified. These mutations include the transfer of a nonsense mutation to codon 318, which causes the salt-wasting form of CAH; an isoleucine-to-asparagine change, which cause the simple virilizing form; an eight-base pair deletion, which produces a nonfunctional protein because of a frame shift that causes the salt-wasting form of the disease; and a conversion of the sixth intron to part of the seventh exon, which causes the late-onset form. A triple substitution of tyrosine for adenosine in the sixth exon occurs with equal frequency in all three forms of CAH21.14
In addition, random point mutations that change single amino acids may cause abnormal protein function of the CYP-21B gene product. For instance, the late-onset form of CAH21 is associated with a guanosine-to-cytosine substitution in the seventh exon.14
CLINICAL PRESENTATION.
Depending on the severity of the P45021 defect, the presentation of CAH can vary from the severe classic virilizing form to the late-onset form, in which hirsutism develops at puberty, to the cryptic (asymptomatic) form, which can be detected only by genetic linkage studies or provocative hormonal testing.18 In the classic form, the female neonate displays profoundly virilized external genitalia, include clitoromegaly and labial fusion due to prenatal elevation of the undulating levels of testosterone (T) and dihydrotestosterone (DHT). However, the ductal structures are female in phenotype, featuring normal cervix, uterus, fallopian tubes, and ovaries. In contrast, boys with this disorder display normal external genitalia at birth. However, both sexes are affected by the high circulating levels of androgens manifested by precocious puberty and early fusion of the epiphyses, resulting in short stature.
In 75% of the patients with classic CAH21, severe aldosterone deficiency leads to sodium depletion.19 This salt-wasting form of CAH21 can be attributed not only to cortisol deficiency but also to the enzymatic defect at the level of the zona glomerulosa, where the sodium-retaining steroid aldosterone is the major product. In the simple virilizing form, the enzyme functions abnormally only in the zona fasciculata that is responsible for cortisol production.
The patients with the late-onset form of CAH21 have varying degrees of precocious puberty, hirsutism, cystic acne, clitoromegally, short stature, male pattern baldness, anovulation, oligomenorrhea, and infertility.2,20,21 Virilization is rare. The frequency of late-onset CAH21 deficiency in hirsute women ranges from 0% to 30%.22 Hirsute patients diagnosed with CAH were more likely to have severe hirsutism, virilization, short stature, family history of hirsutism, earlier onset of symptoms, and regular menses. They also may have a condition similar to polycystic ovary syndrome (PCO), with anovulation and hirsutism. Four percent of patients diagnosed with PCO tested by ACTH stimulation had late-onset CAH21.23
INCIDENCE.
Heel capillary screening of newborns has been performed with a 17OHP assay with a microfilter paper method that was developed by Pang and associates in 1977 and has been used for population screening.24 As calculated by worldwide screening of newborns, there is a 1:60 incidence of heterozygous carriers with a gene frequency of 0.0082. This finding excluded studies in two populations in which the gene frequency was elevated (the Yupik Eskimos of southwest Alaska and the population of La Reunion, France). Infants who had clinical or biochemical evidence of CAH21, such as hyponatremia (serum sodium level less than 130 mEq/L) with associated hyperkalemia (>6.5 mEq/L) or acidosis (HCO3 < 17 mEq/L) were classified as having the salt-wasting form. With these criteria, the incidence of the salt-wasting form is three times higher than that of the simple virilizing form. Premature, severely ill, and low-birth-weight infants had a higher false-positive rate in the screening program.19
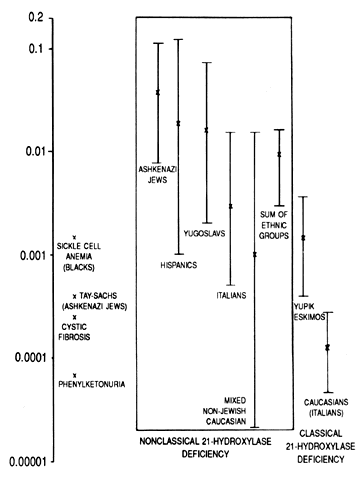
Obviously, neonatal screening programs cannot determine the incidence of late-onset CAH21 because the baseline circulating hormone levels of these patients are normal. By studying the haplotypes of the parents of affected persons and determining whether they are heterozygous or homozygous for 21-OH deficiency by hormonal studies, the incidence of the occurrence of late-onset genes was determined in several populations. Ashkenazi Jews (3.7%), Hispanics (1.9%), Yugoslavs (1.6%), and Italians (0.3%) have a high incidence of attenuated CAH21. In a mixed group of white persons, only 0.1% of affected subjects were homozygous. With sib-pair analysis rather than parental analysis, a similar result was obtained (Fig. 2).25
|
HORMONAL DIAGNOSIS.
Classic CAH21 is diagnosed by documenting basally elevated circulating levels of 17OHP. The circulating levels of dehydroepiandrosterone (DHEA), androstenedione, and T26 also are elevated because hormone production is shifted toward androgen biosynthesis. Interestingly, the circulating levels of pregnenolone and 17α-hydroxypregnenolone also can be elevated in classic CAH21; an increase can be explained by either oversaturation of the capacity of 3β-HSD or inhibition of 3β-HSD by the accumulation of its products.27,28 These precursors are converted to progesterone and 17OHP by the steroidogenic enzyme 3β-HSD.
In late-onset CAH21, basal endocrine determinations may or may not show an increase in the circulating levels of progesterone, 17OHP, androstenedione, T, DHEA, dehydroepiandrosterone sulfate (DHEAS), 11-desoxycortisol, or combinations thereof.27 Significantly, the variable occurrence of basal elevations of these hormones is in large part due to their diurnal (and episodic) fluctuation. Consequently, the timing and frequency of sampling may determine the productivity of basal endocrine testing.
In view of the variable utility of basal endocrine testing, standardized ACTH stimulation tests have been used in recent years.28,29,30,31,32 These tests usually involve the measurement of steroid precursors proximal to the enzymatic block before and after ACTH stimulation. This approach is based on the premise that subtle enzymatic deficiencies may be brought to light under circumstances that challenge the adrenal steroidogenic machinery. Although in some cases dexamethasone preparation the night before testing has been used, current experience indicates that dexamethasone preparation does not alter corticoid and androgen responses to ACTH.33 The simpler diagnostic test described by Gutai and colleagues30 does not involve dexamethasone preparation. The test assesses the combined rate of increase of progesterone and 17OHP levels over a 30-minute testing period (Fig. 3). A value of greater than 6.5 ng/dl/minute is abnormal.
|

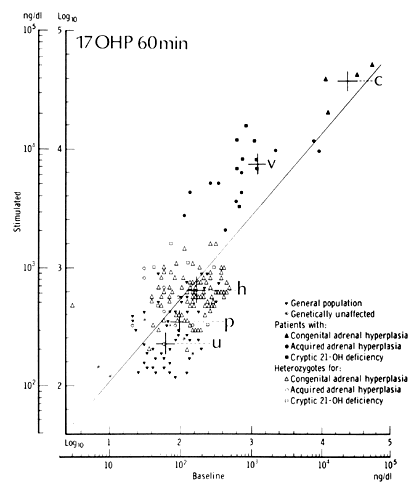
It is necessary to test female patients during the follicular phase, thereby avoiding the confounding effect of the luteal rise in the circulating levels of progesterone. By measuring 17OHP 60 minutes after administration of 2.5 mg ACTH, New34 developed nomograms to distinguish patients with classic, late-onset, asymptomatic, and heterozygous CAH21 from a control population (Fig. 4). The high incidence of disease in the control population may account for the overlap in values in the nomograms. Elevations in ACTH-stimulated androstenedione level and 17OHP:11-DOC ratio confirm the diagnosis.35 However, if a baseline 17OHP value is greater than 500 ng/dl, late-onset CAH21 can be confirmed without further testing. Some recommend ACTH testing only when a morning 17OHP level is greater than 6 nmol/L.36,37 Others have shown that basal endocrine determinations cannot identify patients with late-onset CAH21.38
|
PRENATAL DIAGNOSIS.
Prenatal diagnosis of classic CAH21 allows for early treatment or abortion of an affected fetus and reassurance of the parents of unaffected fetuses. It was first determined in 1975 that 17OHP levels measured in the amniotic fluid could predict classic CAH21 at 24 to 28 weeks.39 Therefore, second-trimester diagnosis can be made by amniocentesis. Control values differ significantly between laboratories; therefore, each laboratory must standardize its own values. Amniotic fluid levels of androstenedione and 21-deoxycortisol also are elevated and T is in the normal male range in female fetuses with CAH21. Steroid analysis of amniotic fluid cannot be used to diagnose the late-onset form, heterozygotes, or prenatally treated fetuses because amniotic fluid hormone levels are normal in these conditions.40,41,42,43
When the genetic linkage of CAH21 to HLA genes was acknowledged, methods were developed to perform HLA typing on the amniotic fluid cells to compare them with an affected sibling HLA type and the parental HLA type.41,42,43,44,45 HLA typing requires polymorphic differences between the index case and each parent to identify which haplotypes in the parents are responsible for the transmission of an abnormal gene. Sometimes, there is not enough difference in the parent haplotype to determine which haplotype is abnormal; in these cases, HLA typing is nondiagnostic. Errors in predictions for CAH21 can be attributed to recombinations, amniotic fluid contamination with maternal lines, and incorrect HLA typing. Predictions were correct by HLA typing and steroid analysis in 85% of cases.45
Mornet and colleagues46 developed DNA probes to HLA genes located in the class I and class II regions that can be used to characterize a fetal genotype expressed on cells obtained by chorionic villus sampling at 10 weeks. This genotype can be compared with maternal, paternal, and index case (a previously affected sibling) restriction fragment patterns. The risk of recombination occurring between these probes and the CYP-21 genes was calculated at 1%. After the CYP-21 genes were sequenced,10,47 a cDNA probe was developed that can identify some deletions in the gene by the absence of a 3.7-kb band that is present in normal persons when Taq1 is used as a restriction endonuclease.48 The correct diagnosis rate with this cDNA probe is 98% to 99% when the electrophoretic band sizes that are obtained with Hind 3, ECO RI, BglII, or TaqI restriction endonucleases are compared with expected band sizes. This method can be performed earlier than HLA typing because it does not rely on gene expression, but the actual DNA.
3β-Hydroxysteroid Dehydrogenase Deficiency
Although less common than CAH21, CAH3β-HSD has similar presentations. A rare salt-wasting classic form of CAH3β-HSD was described in 1962.49 In male infants, incomplete masculinization usually is present because of inadequate testicular production of testosterone and other androgens. On the other hand, hirsutism in women with late-onset CAH3β-HSD results from the peripheral conversion of the Δ5 steroids to Δ4 steroids in situ at the target organ level. An allelic variant that causes a late-onset (pubertal, attenuated) form is becoming more commonly recognized in patients who are hirsute, have acne, and have a PCO-like condition.50 Some patients have precocious puberty, menstrual irregularities, or varying degrees of virilism. The estimated incidence of CAH3β-HSD in hirsute women appears to be 12.9%, or approximately one in eight hirsute women.51 Lucky and associates52 studied a group of women with acne and found CAH3β-HSD in 10%.
The 3β-HSD enzyme converts Δ5 steroids to Δ4 steroids in both the ovary and adrenal gland. Therefore, unlike CAH21, CAH3β-HSD affects both organs. 3β-HSD is not a cytochrome P450 protein and is not HLA linked. It recently has been located on chromosome 1. Type 1 and 2 species have been identified with equal substrate affinity. Type 1 3β-HSD is expressed in human skin, mammary glands, and adipose tissue, whereas type 2 3β-HSD is expressed in the ovary and adrenal gland.53
Most patients with classic CAH3β-HSD have the salt-wasting form, but a few have normal aldosterone production. There is a 3β-HSD defect in the 17-deoxysteroid pathway in the zona fasciculata in both classic forms, but in the salt-wasting form, the 17α-hydroxysteroid function also is nonfunctional in the zona glomerulosa. Patients with classic CAH3β-HSD have elevated basal and ACTH-stimulated pregnenolone, 17α-hydroxypregnenolone, and DHEA levels. Unexpectedly, Δ4 steroids that are produced past the enzyme block also have slightly elevated levels. However, the ratio of Δ5 to Δ4 steroids is greatly increased. The conversion of DHEA to androstenedione, testosterone, and DHT in the skin and hair follicles was shown previously.54,55,56 It is proposed that the elevation of Δ5 to Δ4 steroids is caused by peripheral and liver conversion, which shows that the genetic defect may occur in the type 2 mRNA 3β-HSD transcript that is expressed in the ovary and adrenal gland, whereas a normally functioning type 1 mRNA 3β-HSD transcript is expressed in peripheral tissues.53
Usually, ACTH stimulation is needed to determine the abnormal hormone function in late-onset CAH3β-HSD. The baseline ratio of circulating 17α-hydroxypregnenolone to 1/OHP was higher in 20 hirsute patients with late-onset CAH3β-HSD (greater than 2.8) than in control subjects (1.2). After ACTH stimulation, this ratio increased to 8 to 14 in the patients with late-onset CAH3β-HSD compared with 4.4 in normal women and less than 0.5 in patients with CAH21. Despite the elevation of serum Δ4 steroids (androstenedione and T), the ratio of DHEA to androstenedione in women with attenuated CAH3β-HSD was significantly higher than that of normal women, usually more than 8.50,51 In contrast, ACTH-stimulated circulating levels of 17OHP, androstenedione, corticosterone, and 11-DOC in these women did not differ significantly from those of normal women, suggesting that 11β-hydroxylase activity was normal.
In women with PCO, serum 17α-hydroxypregnenolone and DHEA levels may be moderately elevated compared with control subjects. After ACTH stimulation, however, the serum levels of these steroids were comparable to those of normal women. Thus, women with attenuated CAH3β-HSD can be distinguished from women with PCO by the observation that the level of Δ5 steroids in the latter group may not be as high in either the basal or ACTH-stimulated state. In addition, the elevations of serum androstenedione and T in women with late-onset CAH3β-HSD were at least partially dexamethasone suppressible, suggesting both an ovarian and an adrenal origin. In contrast, the elevated levels of serum androstenedione and T in women with PCO were not dexamethasone suppressible, suggesting that these androgens are of ovarian origin.57
In summary, ACTH stimulation resulted in exaggerated ratios of 3β-HSD precursor to product for both C21 and C19 steroids. The most strikingly increased ratio is 17α-hydroxypregnenolone to 17OHP. Significant elevation of the ratio of DHEA to androstenedione also has been observed. As in the case of late-onset CAH21, these patients often had a clinical picture that was indistinguishable from that of patients with PCO.57 The ACTH stimulability, the dexamethasone suppressibility, and the characteristic circadian rhythm of the elevated Δ5 steroid levels are interpreted as evidence of the adrenal origin of attenuated CAH3β-HSD. Radiologic evidence of bilateral adrenal hyperplasia also has been reported. Hirsutism in patients with late-onset CAH3β-HSD is presumed to be caused by increased circulating levels of andostenediol and free T caused by peripheral conversion.
11β-Hydroxylase Deficiency
Fewer than 10% of patients with CAH have CAH11β. It has allelic variants that cause classic and late-onset forms. Some patients have hypertension because of elevation of the circulating levels of 11-DOC (precursor at blocked enzyme), which promotes sodium retention, plasma volume expansion, and suppressed plasma renin activity. Other factors may contribute to the hypertension because normalization of the 11-DOC level does not always rectify the hypertension.2
The gene for 11β-hyroxylase (CYP-11) is located on chromosome 8q, along with a homologous gene of unknown function. Therefore, it is not linked to the HLA system, but is located near MYC, NOS, and the thyroglobulin gene.58 The protein encoded is a cytochrome P450 that functions in the mitochondria. It performs several enzyme functions: 11β-hydroxylase activity, 18-hydroxylase activity (corticosterone methyl oxidase I[CMO-I] activity), and 18-dehydrogenase activity (corticosterone methyl oxidase II[CMO-II] activity). The last function is performed only in the zona glomerulosa. CMO-II deficiency is an allelic variant that results from mutations in the CYP-11 structural gene.59
A high rate of CAH11β is found in Moroccan and Turkish Jews. Many of these families have a single base change in the CYP-11 that causes a substitution of histidine for arginine, which is conserved in all known eukaryotic P450 proteins.60
The clinical presentation of patients with CAH11β is indistinguishable from that of patients with CAH21, with the notable exception of the coexistence of systemic hypertension. Basal testing typically shows elevated circulating levels of 11-DOC as well as progesterone, 17OHP, and C19 androgens in the classic form.
Diagnosis of late-onset CAH11β is established by an elevated ratio of 18-hydroxycorticosterone to aldosterone in the serum. Maroulis and associates61 evaluated the possible occurrence of CAH11β in a population of hirsute patients and concluded that it is not a common cause of hirsutism. However, several other investigators maintain that appropriate evaluation of this possibility will result in a measurable incidence of CAH11β in hyperandrogenic patient populations.62,63,64,65,66 Lucky and colleagues52 found evidence of CAH11β in 2 of 31 patients with acne.
Prenatal diagnosis of classic CAH11β can be made by detection of elevated levels of 11-DOC67 in amniotic fluid or elevated maternal levels of urinary tetrahydrodeoxycortisol (THS) after 8 weeks of gestation. THS is a urinary metabolite of 11-DOC that is not detected in the urine of normal pregnant women but increases progressively in pregnant women with a fetus affected by CAH11 β.68
20,22-Desmolase Deficiency (Congenital Lipoid Adrenal Hyperplasia)
Deficiency of 20,22-desmolase (P450SCC) is a rare, usually fatal disorder in which the first step in steroidogenesis is blocked. Side chain cleavage of cholesterol to pregnenolone cannot be performed by the cells of affected infants. Regardless of the genotype, all infants are phenotypic females. Profound adrenal insufficiency is present. At autopsy, the adrenal glands are large and filled with lipid.69
17α-Hydroxylase Deficiency
A rare form of congenital adrenal hyperplasia, 17α-hydroxylase deficiency usually is diagnosed around the time of puberty. The involved P450 enzyme that catalyzes 17α-hydroxylase and 17, 20-lyase reactions is not necessary for mineralocorticoid synthesis, but is essential for the formation of cortisol, androgens, and estrogens.70
The CYP-17 gene is located on chromosome 10. Four allelic variants that result in nonfunctional P45017 enzyme have been identified. They include a duplication, a substitution, and two deletions.71 At puberty, these patients are sexually immature girls with hypertension and hypokalemia. The high levels of ACTH increase the synthesis of 11-DOC and corticosterone, and produce sodium retention and potassium depletion. Although aldosterone synthesis is unimpaired, the secretion of aldosterone is decreased because the renin-angiotensin system is depressed. Plasma cortisol and gonadotropin levels are elevated. In general, the hormonal picture is the same in male and female patients.
Treatment of Congenital Adrenal Hyperplasia
Treatment consists of providing replacement levels of glucocorticoids.4 In the classic form, where there is complete enzyme blockage, it is reasoned that providing the equivalent of the daily secretion rate of cortisol by exogenous means will reduce the excessive secretion of ACTH to levels normally observed under physiologic concentrations. As a result, ACTH-driven adrenal androgen production should diminish, and endogenous cortisol secretion should stop. The total daily dose administered should provide the equivalent of the daily cortisol secretion rate to mimic, as much as possible, physiologic cortisol secretion and achieve homeostatic regulation of ACTH release.
The daily cortisol secretion rate in normal adults averages 12.5 ± 2 mg/m2/day or 15 to 20 mg/day.72 However, if cortisol is given orally, in the form of hydrocortisone tablets, the overall replacement dose may have to be at least doubled because a significant portion of the drug is likely to be inactivated by gastric acidity. Therefore, the daily maintenance dose of oral cortisol is approximately 25 mg/m2/day.73 For a woman of average size (height 5'5” [165 cm], weight 110 lb [50 kg]), the calculated surface area of 1.5 m2 would indicate a daily replacement dose of hydrocortisone of 37.5 mg/day.
If a synthetic glucocorticoid (e.g., dexamethasone, prednisone) is used, several additional factors come into play, complicating the calculation of the optimal replacement dose. Primarily, there is no reliable database with which to compute the daily replacement dose of a synthetic glucocorticoid preparation that would be equivalent to cortisol 37.5 mg/day. Reports of the relative potencies of a variety of glucocorticoid preparations vary widely. For example, dexamethasone has been estimated to have a glucocorticoid effect 25 to 100 times more potent than that of hydrocortisone. Clearly, such variable estimates complicate the decision about the optimal replacement dose. Although all glucocorticoids can induce pseudotumor cerebri, that tendency appears to be exaggerated with some of the synthetic preparations. Nevertheless, given the cumulative clinical experience, a total daily dose of dexamethasone 0.5 to 0.75 mg usually is adequate for the task. Nightly administration for the inhibition of the nocturnal ACTH surge is desirable.74 Likewise, total daily doses of prednisone of 5 to 7.5 mg probably are equally effective.
Patients must be observed to ensure that the levels of adrenal androgens have been reduced. It is particularly helpful to follow the steroid that is most proximal to the enzymatic block that tends to accumulate in the entity in question (Table 1).75 The regimen given above is not intended for total pituitary adrenal suppression.75,76,77,78 Nevertheless, some patients may be more sensitive than others to the effects of exogenous glucocorticoids, and they may experience undesirable significant suppression of the pituitary adrenal axis. The ACTH-adrenal axis of these patients may not be able to respond to stress in the form of accident, surgery, or infection, and temporary augmentation of the exogenous glucocorticoid dose may be needed to prevent adrenal crisis. Patients who are taking exogenous glucocorticoids should follow the precautions described for Addison's disease. However, patients treated according to this regimen are not likely to have complete adrenal insufficiency or to require such extreme measures.79,80 To test for this possibility, a standard ACTH test can be performed.81 If the cortisol response is normal, the patient and physician do not need to be concerned. However, if the ACTH stimulation test result shows complete suppression, the physician should consider lowering the dose. In some patients, complete pituitary adrenal suppression is necessary to achieve control. Under those circumstances, the standard precautions used for patients with Addison's disease should be followed. According to some investigators, suppression of morning basal cortisol levels to less than 2 μg/dl is excessive and requires dose modification.
Enzyme Defect | Elevated Plasma Steroid Level | Elevated Urinary Steroid Level |
21-hydroxylase deficiency | 17-hydroxyprogesterone | Pregnane-3α, 17α, 20α-triol |
| (17-OHP) | (pregnanetriol) |
|
| pregnane-3α, 17α, 20α-triol-11-one |
|
| (pregnanetriolone) |
|
| 17-hydroxypregnanolone |
11-hydroxylase deficiency | 11-desoxycortisol (S) | tetrahydro-11-desoxycortisol |
|
| (THS) |
3β-hydroxysteroid dehydrogenase | Δ5-pregnene-3β-ol, 20-one | Δ5-pregnene-3β, 17α, 20α-triol |
deficiency | (Δ5-pregnenolone) | (Δ5-pregnenetriol) |
| dehydroepiandrosterone |
|
| (DHEA) |
|
A potent mineralocorticoid, 9α-fluorohydrocortisone or fludrocortisone (Florinef, Squibb Mark, Princeton, NJ; 150 μg/m2/day, 0.2 mg/day), should be started when salt wasting develops in infants with CAH. Salt supplementation of 2.2 mmol/kg/24 hours may be required in the acute phase. Plasma renin levels should be observed to assure that they are normal. Excessively large amounts of hydrocortisone may be needed if fludrocortisone is insufficient.82 Inadequate replacement can cause failure to thrive.
Extensive clinical experience shows that suppression results in the resumption of menstrual cycles in anovulatory patients.4,73,83 Although the reason for the improvement in ovulatory status remains uncertain, a reduction of the total effect of androgen, perhaps at the central as well as the intraovarian level, may be responsible. Clearly, androgens may exert a negative feedback effect on gonadotropin release and also may arrest follicular development. If adequately treated, patients with classic CAH will have normal pubertal progression.73
Although late-onset CAH is thought to be best treated by glucocorticoid replacement, a recent study suggested that treatment with cyproterone acetate, a peripheral androgen inhibitor, provides greater clinical improvement in hirsutism scores despite continued abnormalities in androgen profiles.84
Prenatal treatment of female fetuses was encouraged by David and Forest,85 who noted less virilization in females who had CAH21 and had been treated with dexamethasone than in their older, untreated affected sisters. The basis of prenatal treatment presupposes that the fetus has an intact pituitary adrenal axis controlled by ACTH. Gestational treatment with both dexamethasone and hydrocortisone has been attempted.86,87,88,89 Dexamethasone is the drug of choice because its transplacental passage is far greater than that of cortisol.88 Marked suppression of the adrenal gland was shown after treatment of affected female fetuses with dexamethasone as assessed by the low levels of estriol (fetal androgen metabolite) in the amniotic fluid and maternal urine.89 A dose of 20 μg/kg prepregnancy weight, provided in three equal doses, is recommended (approximately 5 mg/three times a day). Low-dose dexamethasone treatment during pregnancy was not teratogenic.2,87,88