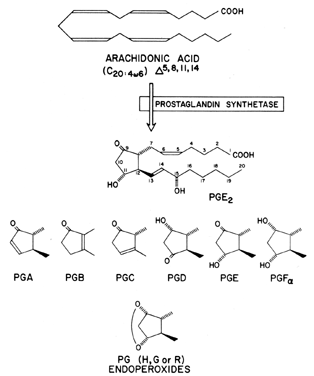
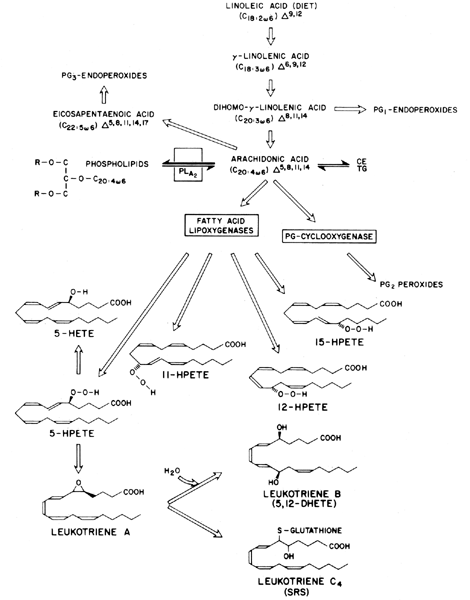
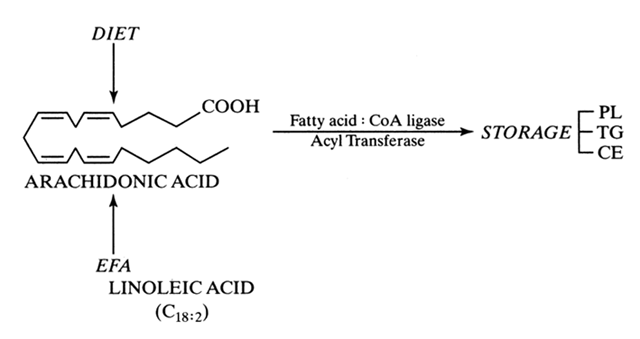
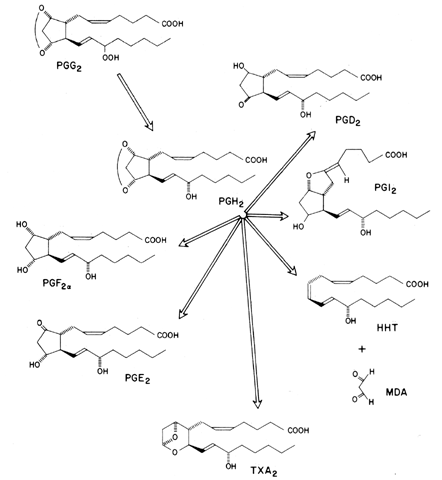
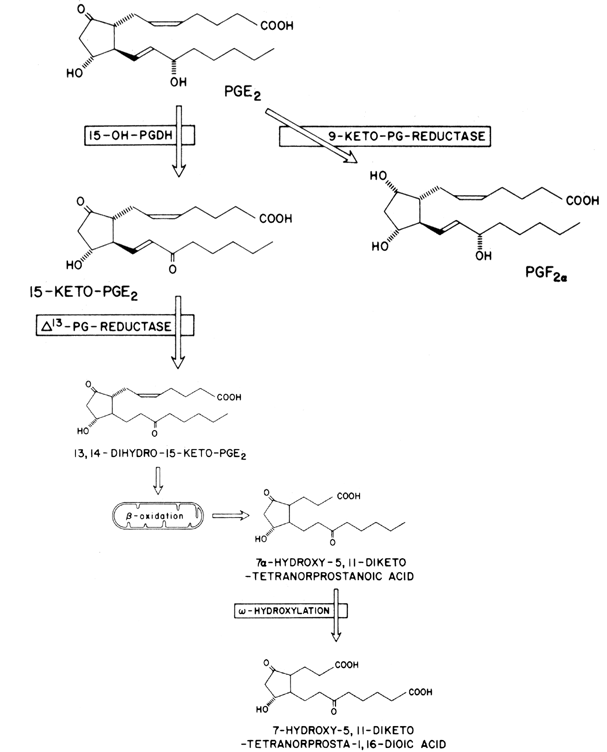
Prostaglandins exhibit a wide range of biological effects, and their actions
are among the most varied of any naturally occurring compounds. Despite
this observation, this group of lipids displays a marked structure-activity
specificity, which is determined mainly by cyclopentanone
ring substitutions and the degree of unsaturation of the prostanoic
acid side chains. The cellular response to prostaglandins is mediated
by their interaction with plasma membrane receptors. PG receptors were initially classified on the basis of functional activities
of natural and synthetic agonists, and antagonists were classified
into the following categories: DP, EP, FP, IP, and TP. The first letter
denotes the prostaglandin type, and the letter P stands for “prostanoid.”30 Later, studies by binding analysis and molecular cloning confirmed the
presence of distinct receptor types as well as three or four subtypes
of EP (EP1–4). Plasma membrane prostaglandin receptors belong to the superfamily of
G-protein-coupled receptors characterized by seven transmembrane-spanning
regions.31 Intracellular second messengers of prostaglandin receptors include cAMP, protein
kinase C, and calcium.30,31 The various receptors show remarkable specificity for the eicosanoid, with
at least a 100-fold preference for the ligand. At high concentrations, PGE2 and PGF2α interact with the DP receptor; similarly, PGF2α will activate the EP receptor, whereas PGD2 and PGE2 will interact with the FP receptor at high concentrations. Most tissues
contain a mixture of receptors, which appears to be the basis for the
often opposite effects of a particular prostaglandin at different doses. Role of Prostaglandins in Gonadotropin Secretion Recent data implicating a physiologic role of prostaglandins in the regulation
of gonadotropin-releasing hormone (GnRH) secretion have been published.32,33 A number of reports now suggest that prostaglandins, particularly PGE2, exert stimulatory influences on gonadotropin release, an effect that
appears to be mediated by an action at the level of the hypothalamus. PGE2 has been shown to stimulate the release of GnRH from the hypothalamus, and
pretreatment of animals with antisera to neutralize endogeneous GnRH
prevents the PGE2-induced release of LH.34 In addition, PGE1 and PGE2 appear to be the most potent stimulators of growth hormone release from
cultured adenohypophyseal cells.35 PGB, PGA1, PGA2, PGB2, PGF1, and PGF2 are also active stimulators, but their effects are not seen at physiologic
doses. In general, prostaglandins appear not to stimulate gonadotropin
secretion by a direct action on the pituitary. Further evidence suggesting that prostaglandins might affect gonadotropin
secretion (by acting directly on the hypothalamus) is supplied from
studies using prostaglandin synthetase inhibitors, such as indomethacin, which
apparently reduce gonadotropin secretion.36 That prostaglandins act by way of hypothalamic-releasing factors is evident
on the basis of two observations: - Pretreatment of female and male rats with antiserum to LH-releasing hormone (LHRH) impaired
the ability of PGE2 (100 mg per rat) to increase LH secretion.36
- Direct administration of PGE2 into the ventricle of the brain of the rat mimicked the intravenous effect
of prostaglandins on the stimulation of gonadotropin secretion.36
Additional findings provide compelling support for the concept that PGE2 acts at the level of the median eminence to elicit release of LHRH. This
contention is further corroborated by the finding that median eminence
tissue contains greater amounts of endogenous prostaglandins than
basal hypothalamic regions. Data obtained from in vitro incubations of
median eminence fragments obtained from male rats have shown that norepinephrine
and dopamine stimulate the simultaneous release of LHRH and
PGF2α. This effect is blocked by indomethacin, suggesting that intraneuronally
produced prostaglandins are the mediators of catecholamine-stimulated
LHRH release. These interesting findings are discussed in greater detail
in a review by Ojeda and co-workers.36 Ovulation and Prostaglandins After the discovery that indomethacin and aspirin (inhibitors of prostaglandin
synthesis) could block ovulation,33,37 it was suggested that prostaglandins were involved in the ovarian follicular
rupture process. This contention was further strengthened by the
finding that intraovarian injection of PGF2α antiserum also inhibited ovulation.38 There is now a substantial amount of evidence indicating that follicular
prostaglandin formation is enhanced during ovulation and that this
elevation is dependent on gonadotropins.39 The midcycle surge of gonadotropins stimulates follicular eicosanoid biosynthesis
by a cAMP-mediated process that is dependent on gene activation, but
independent of steroidogenesis. LH appears to be the dominant
physiologic pituitary gonadotropin responsible for the induction of
ovulation, and it seems likely that the effects of LH on follicular rupture
may be mediated by leukocytes that secrete proteolytic enzymes, oxygen
radicals, and prostaglandins. Indomethacin, for instance, will
block ovulation normally induced by large doses of human chorionic gonadotropin
in vivo. Prostaglandins may mediate the stimulatory effects
of LH on “ovulatory enzymes,” such as protease or collagenase.40 There is also the possibility that prostaglandins may elicit a contractile
response in the follicle wall,41 which is now known to contain contractile elements, such as myosin and
actin. Plasminogen activator or some other protease appears to be intrinsically
involved in follicle rupture,42 and it is evident that secretion of this protein is associated with the
LH-induced rise in follicular prostaglandin biosynthesis, although it
has been pointed out that these two events may not be interdependent. A
possible mechanism of prostaglandin action in follicular rupture is
shown in Figure 6. 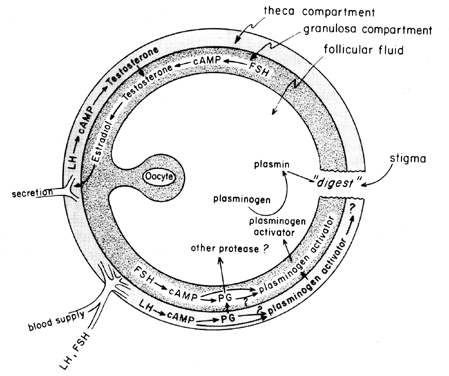 Fig. 6. Possible mechanism of prostaglandin action in follicular rupture.(Behrman HR: Prostaglandins in hypothalamo-pituitary and ovarian function. Ann
Rev Physiol 41:685, 1979) Fig. 6. Possible mechanism of prostaglandin action in follicular rupture.(Behrman HR: Prostaglandins in hypothalamo-pituitary and ovarian function. Ann
Rev Physiol 41:685, 1979)
|
Role of Prostaglandins in Luteal Function: Luteolysis and Menstruation The mechanism by which the human corpus luteum regresses 10 to 12 days
after its formation is a mystery. Since the early finding that PGF2α was luteolytic in the rat and many other subprimate species,43 a major research effort has been made to investigate the possibility of
menstrual regulation with PGF2α.PGF2α induces functional regression of the corpus luteum by a receptor-mediated
process, independent initially of changes in ovarian or luteal blood
flow.32 Within minutes, PGF2α depletes ascorbic acid,44 uncouples the occupied LH receptor from adenylate cyclase, and decreases
transport of gonadotropin from capillaries to the luteal cell. A review of the pertinent literature reveals many conflicting reports on
the effects of PGF2α in humans. Some studies have recorded transient declines in circulating
levels of progesterone by PGF2α,45 although other studies have failed to demonstrate such an effect in normally
ovulating women.46,47 In view of the existence of putative PGF2α receptors in the human corpus luteum,48 it was reasoned that the failure to achieve luteolysis with intravenous
infusions of PGF2α was perhaps due to rapid pulmonary metabolism. Subsequent studies involving
the direct injection of PGF2α into the human corpus luteum induced progesterone withdrawal, the nadir
of which coincided with the onset of early menses.49 These findings suggest that endogenous ovarian prostaglandins (or some
other luteolytic agent) may be required for luteal regression. This seems
feasible in view of the observation that human luteal function appears
to be independent of uterine control, as neither hysterectomy50 nor congenital absence of the uterus, vagina, or fallopian tubes affects
ovarian cyclicity.51 Human luteal tissue is certainly capable of producing prostaglandins, as
shown by in vitro experiments52; however, attempts to correlate increased PGF2α levels in the ovary with luteal regression in the late luteal phase have
not been successful.53 It seems that the only time ovarian PGF2α production is enhanced in the human menstrual cycle is shortly after ovulation.54 It is also apparent, however, that concentrations of PGF2α are always higher in ovarian stromal tissue through early, mid, and late
luteal phases. In contrast to the in vivo effects of PGF2α, in vitro studies have demonstrated both transient and sustained luteolytic
effects in human luteal tissue.55,56,57 It is the general consensus that an early event in PGF2α-induced luteolysis is the abrogation of gonadotropin-sensitive cAMP production
derived from studies in the rat corpus luteum.58,59 This abrogation is likely to be by a direct inhibition of adenyl cyclase
by increases in intracellular Ca2+.60 Recent evidence from the field of neuroscience indicates that prostaglandins
stimulate the release of intracellular Ca2+ by increasing the hydrolysis of phosphoinositol to diacylglycerol and
inositol triphosphate.61 Consistent with findings in laboratory animals, it was shown that regression
of the human corpus luteum is also associated with a loss of LH
receptors.62 It is well known that the newly formed corpus luteum of many species is
refractory to the lytic action of PGF2α63; this observation also appears to extend to the human corpus luteum.64 Henderson and McNatty65 suggested that PGF2α receptors in the newly formed corpus luteum may somehow be masked by occupied
LH receptors, and that gradual dissociation of LH from its receptor
may increase the susceptibility of the corpus luteum to PGF2α. An alternative hypothesis was offered by other investigators,65 who suggest that the susceptibility of the human corpus luteum to PGF2α may be dependent on ovarian noradrenaline levels, which increase during
the luteal phase. This increase in catecholamines appears to permit
the antigonadotropic effect of PGF2α on human corpus luteum.66 Prostaglandins in the Uterus Unlike the myometrium, which mainly produces PGI2, the nonpregnant endometrium predominantly produces PGF2α and PGE2. The synthesis of prostaglandins is greater in the glandular epithelium
than in the stroma of the endometrium, and during the secretory phase
than during the proliferative phase of the cycle.67,68 Both PGF2α and PGE2, through their interaction with specific receptors, are known to stimulate
myometrial contractility, leading to an increase in intracellular
Ca2+. Such a mechanism of prostaglandin-induced uterine hypercontractility
has been implicated in the pathogenesis of primary dysmenorrhea. Elevated
levels of PGF2α and PGE2 have been identified in the endometrium and menstrual fluid of women with
dysmenorrhea, and antiprostaglandin agents have been shown to result
in a marked reduction in pain in these patients. Various prostaglandins are also produced in the uterine cervix, and receptor
sites for both PGE and PGF are present in the cervix.69 The increased production of prostaglandins, or their local administration, is
associated with cervical ripening in pregnant women.69 Prostaglandins and Parturition A number of observations indirectly implicate the involvement of prostaglandins
in parturition. It is known, for example, that the administration
of indomethacin or other nonsteroidal anti-inflammatory drugs, such
as aspirin, prolongs gestation.69 Moreover, prostaglandins, particularly PGF2α, are known to be potent stimulators of uterine contractility and induce
cyclic contractions of the gravid uterus. Elevated prostaglandin concentrations
are associated with the onset of spontaneous labor in humans. Furthermore, the
period of time preceding the onset of contractions
is characterized by plasma PGF2α levels that are equivalent to those of nonpregnant women. Immunization
against PGF2α also delays the onset of parturition.70 Prostaglandins are now used to induce early labor and abortion; indeed, prostaglandin
is the drug of choice for accomplishing midtrimester abortions.69,71 Based on extensive studies in sheep, it appears that prostaglandins synthesized
locally in the uterus initiate labor by directly acting on the
myometrium to promote uterine contractions. A decrease in local, placental
progesterone, combined with an increase in estrogen, results in
the release of Ca2+ from lysosomes that have become fragile. This Ca2+ release stimulates prostaglandin synthesis by increasing phospholipase
activity. Oxytocin appears to facilitate, rather than initiate, the estrogen
and progesterone effects.72 Fetal membranes, decidua vera, and myometrium may all participate in the
formation of prostaglandins.73,74 A recent proposal is that prostaglandin production by intrauterine tissue
is tonically inhibited during human pregnancy, and that such inhibition
is progressively removed as term approaches. Although the mechanisms
effecting the tonic inhibition of prostaglandin synthesis have not
been established, several antiphospholipase peptides have been found
in human placenta, amnion,75 and chorion.76 Another possibility is that prostaglandin synthesis is stimulated in parallel
with removal of synthesis inhibition. Potential signals for increased
synthesis of prostaglandins are several growth factors and platelet-activating
factor. Fetal lung and kidney tissues secrete platelet-activating
factor, which is capable of stimulating prostaglandin production
by human amnion.77 Prostaglandins and the Ductus Arteriosus The maintenance of patency (relaxation of vasodilation) of the ductus arteriosus
is pivotal in controlling the oxygenation of tissues in the
fetus. After birth, the ductus arteriosus normally becomes constricted
or loses its patency. The mechanism by which this closure occurs is unclear, although
blood oxygen tension is believed to be an important factor (Fig. 7). The primary function of the ductus arteriosus in the fetus is to maintain
some degree of left-to-right arterial blood shunting, thereby controlling
the amount of venous return to the lungs. Closure of the ductus
at term is an important physiologic process that, if incomplete, leads
to respiratory distress and cyanosis/hypoxia—syndromes that
are responsible for the high morbidity and mortality in many premature
infants suffering from patent ductus arteriosus. For this reason, there
is a good deal of interest in the mechanism(s) of controlling ductus
arteriosus function. This interest is particularly acute in the field
of prostaglandin research, in which there is now a considerable amount
of evidence suggesting the involvement of prostaglandins in controlling
ductus arteriosus patency at term. 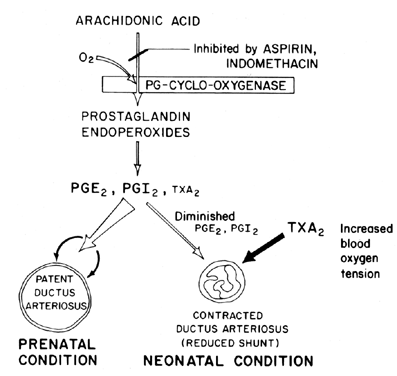 Fig. 7. Scheme for prostaglandin modulation of patency of ductus arteriosus in
fetus and neonate as a function of oxygen tension. Fig. 7. Scheme for prostaglandin modulation of patency of ductus arteriosus in
fetus and neonate as a function of oxygen tension.
|
This evidence is based on the following series of observations: The ductus arteriosus in the fetus with cardiopulmonary deterioration and
hyaline membrane disease can be closed by administration of prostaglandin
synthetase inhibitors.78
Indomethacin can induce contraction of the hypoxic vessel, as demonstrated
by its effects in animals near term.79
Administration of PGE2 (and PGE1) induces relaxation (loss of patency) of isolated fetal lamb ductus arteriosus
preparations (circular strips) in a low oxygen environment (PO2 less than 14 mmHg).80
It seems likely that prostaglandins of the E series, together with prostaglandin
antagonists and prostaglandin-synthetase blockers (aspirin), may
prove to be desirable nonsurgical treatments for preterm infants
with potentially fatal patent ductus arteriosus. Recent evidence suggests
a balance between ductus patency and constriction that is maintained
by synthesis of dilating prostaglandins (PGI2 and PGE2) and constricting prostaglandin (TXA2). In the lamb fetus, both the ductus arteriosus and the lung synthesize
PGI2 and PGE2; then, as term approaches, the lung shifts toward TXA2 synthesis.81 Further information on prostaglandin involvement in the control of ductus
arteriosus function is available.82 |