Maternal Cardiovascular Changes Several cardiovascular changes typically occur during human pregnancy.20,21,22 Heart rate increases by 10% to 15%, cardiac output increases by 30% to 50%, and
blood volume increases by up to 48%. Blood pressure changes
are small in normotensive women, but generally there is a slight decrease
of 2 mm Hg to 3 mm Hg in systolic pressure and 5 mm Hg to 10 mm Hg
in diastolic pressure. Normal blood pressures during the third trimester
average 95/50 mm Hg in the lateral position and 106/65 mm Hg in the
supine position. Women with essential hypertension actually may show
a significant decrease in blood pressure during the first two trimesters. These
facts indicate that normal pregnancy is associated with an
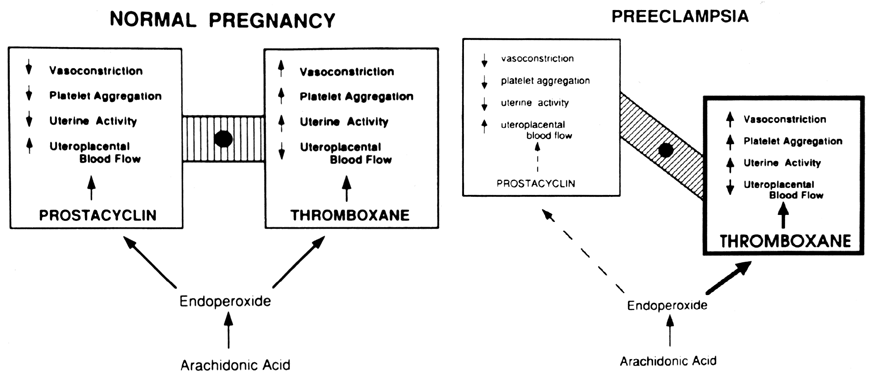
increase in the production of a vasodilatory agent. PROSTAGLANDINS. Prostaglandins are involved in the vasodilatation of pregnancy. Everett
and colleagues23 showed that the dose of angiotensin II (AII) necessary to cause a 20-mm
Hg increase in diastolic blood pressure in pregnant women was significantly
reduced in women treated with indomethacin. Thus, the decreased
sensitivity to the vasoconstrictive effects of AII characteristic of
healthy pregnant women was altered by prostaglandin synthesis inhibition
to a state of increased sensitivity to vasoconstriction. The prostaglandin most likely responsible for increased vasodilatation
in normal pregnancy is prostacyclin (PGI2) because of its potent effect to relax the smooth muscle of blood vessels
and lower systemic arterial blood pressure.24,25,26 Prostacyclin's actions on vascular tone are most likely exerted locally
in the blood vessel wall through a paracrine mechanism between
the endothelial cells, in which prostacyclin is primarily produced, and
the vascular smooth muscle. Leukocytes also can form prostacyclin.27 If prostacyclin is not a circulating hormone, there must be some other
compound that circulates to stimulate prostacyclin synthesis locally in
blood vessels. The identity of such a compound is not known, but it
seems reasonable to assume that the placenta is the source of a circulating
vasodilatory agent. This idea is supported by the facts that blood
pressure typically decreases during pregnancy and that simply turning
a pregnant patient from the lateral to the supine position causes an
increase in blood pressure despite a decrease in cardiac output.21,22 Women in their third trimester (28–32 weeks' gestation) in whom
preeclampsia is destined to develop have a greater increase in blood pressure
than do healthy pregnant women when turned from the lateral to
the supine position.28 One explanation for the increase in blood pressure in the supine position
is that the gravid uterus compresses the vena cava and thereby reduces
venous return to the heart, leading to decreased cardiac output. This
would activate the aortic and carotid sinus baroreceptors to cause
reflex neural vasoconstriction. An alternative explanation is that compression
of the vena cava by the gravid uterus in the supine position
restricts the amount of placentally derived vasodilatory hormone reaching
the maternal systemic arterial blood vessels. THROMBOXANE. The primary source of thromboxane in the adult circulation is the platelets, although
leukocytes also produce it. Thromboxane is a potent stimulator
of platelet aggregation and vasoconstriction,24,25,26 and as such, it has been implicated in several pathologic conditions that
are associated with thrombosis and vascular congestion,25,29 including preeclampsia, as discussed later in this chapter. Under normal conditions, prostacyclin released by the vascular endothelial
cells acts to stimulate an increase in adenosine 3',5'-cyclic
phosphate (cAMP) levels within the platelets, thus inhibiting
aggregation.30 Within the platelets, thromboxane can prevent prostacyclin-induced increases
in cAMP, so under typical circumstances, thromboxane and prostacyclin
counteract each other's actions to maintain homeostasis with
respect to platelet function. If, however, the endothelial cells are
damaged (e.g., injury, atherosclerosis) or prostacyclin synthesis is impaired, then
the actions of thromboxane to produce platelet aggregation and vasoconstriction
are manifested. This mechanism serves to protect the body at
local sites of injury, but in pathologic states in which thromboxane
is allowed to exert its effects throughout an organ or systemically throughout
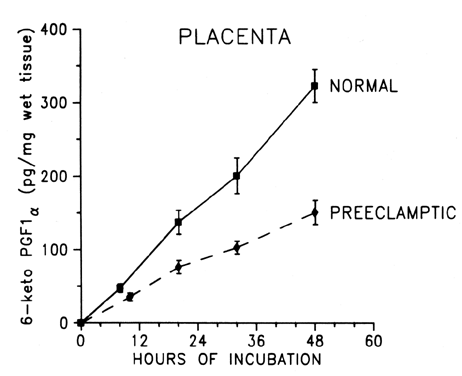
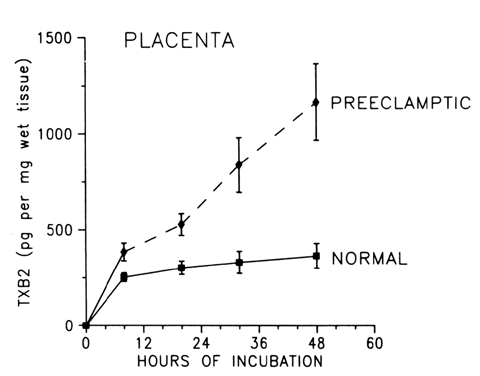
the body, this mechanism can be harmful. During normal pregnancy, the production of thromboxane is increased. Maternal
plasma levels of its stable metabolite, TXB2, as well as its urinary metabolites, are higher during late pregnancy
than during midpregnancy or the nonpregnant state.18,19,31 The placenta, as well as platelets, is an important source of thromboxane
during pregnancy (Fig. 2). Placentas obtained from healthy pregnant women produce approximately 92 μg
of thromboxane daily in vitro,32 and trophoblastic cell cultures, uncontaminated with platelets or other
blood cells, produce 50 to 100 pg of thromboxane per microgram of protein
daily.33 After delivery of the placenta, maternal urinary metabolites of thromboxane
decrease significantly,31 which is a further indication of the placenta as a source of thromboxane
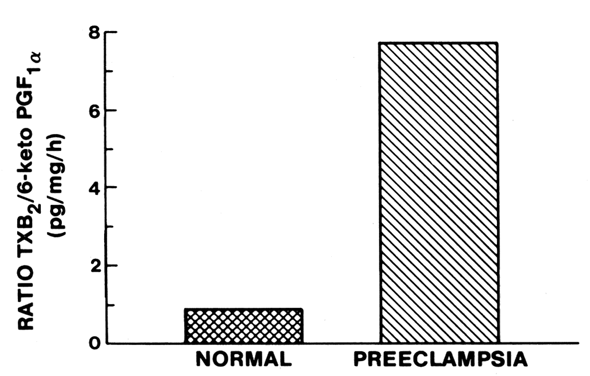
in pregnancy. 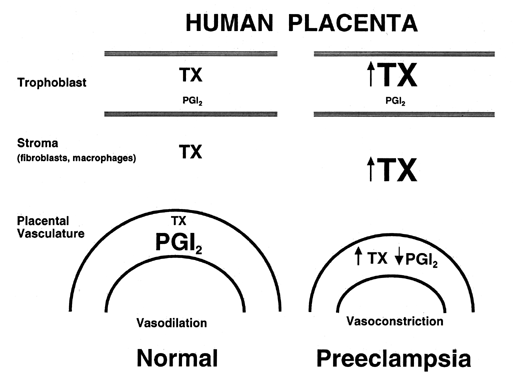 Fig. 2. Compartmentalization of thromboxane (TX) and prostacyclin (PGI2) in the human placenta. TX is produced primarily in the trophoblastic and
stromal tissues, whereas PGI2 is produced primarily in the vascular
tissue. In preeclampsia, TX production is increased coincidentally with
a decrease in PGI2 production. Fig. 2. Compartmentalization of thromboxane (TX) and prostacyclin (PGI2) in the human placenta. TX is produced primarily in the trophoblastic and
stromal tissues, whereas PGI2 is produced primarily in the vascular
tissue. In preeclampsia, TX production is increased coincidentally with
a decrease in PGI2 production.
|
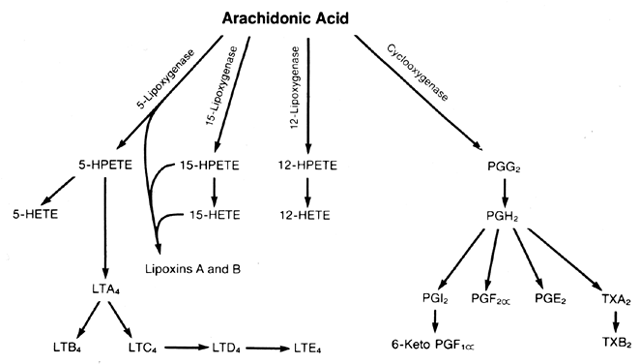
LIPOXYGENASE COMPOUNDS. Arachidonic acid is metabolized by lipoxygenase enzymes in several tissues, organs, and
cells of the body, most notably the lungs and leukocytes (in
which they were first discovered and from which the leukotrienes
derive their name). The human placenta also produces lipoxygenase
compounds. 5-HETE, 12-HETE, 15-HETE, LTB4,16,17,34,35 and LTC4 all have been identified. The physiologic functions of lipoxygenase metabolites during pregnancy
are not known, but the HETEs, HPETEs, lipoxins, and leukotrienes do exert
biologic actions, some of which must be important to placental function
and pregnancy. Both 5-HETE and LTB4 stimulate leukocytic chemotaxis and chemokinesis, increase aggregation
of leukocytes, stimulate the uptake of calcium and d-glucose by neutrophils
and eosinophils, increase bronchial constriction and mucus secretion, increase
vasodilatation, and enhance vascular permeability.36,37,38 LTC4 and LTD4 exert biologic actions in various tissues of the body as reviewed in several
studies.2,37,38,39,40,41 They produce contraction of a number of smooth muscle tissues, including
the small airways of the lung, the ileum, the stomach, the gallbladder, and
the uterus. In the cardiovascular system, leukotrienes often produce opposite effects
in different vascular beds and in different species.2,40,42 For example, LTC4 and LTD4 produce vasoconstriction and cause plasma leakage in guinea pig skin and
the hamster cheek pouch, but they are potent vasodilators in human
skin. LTC4 and LTD4 are potent vasoconstrictors in the coronary circulation of several species. Systemically, LTC4 and LTD4 lower arterial blood pressure in most adult nonpregnant species,2,40,42,43,44 including humans.45 In pregnant and postpartum rhesus monkeys,16,17,46 injection of LTB4, LTC4, or LTD4 (0.5 μg/kg) into the lower vena cava to mimic the systemic route of
placentally produced hormones lowers both systolic and diastolic blood
pressures. The mechanisms of action through which systematically administered leukotrienes
lower blood pressure are not known, but it is likely that they
decrease cardiac output and stimulate prostacyclin production by endothelial
cells in the lungs or locally in the systemic vasculature. LTC4 promotes prostacyclin synthesis by human endothelial cells,47,48 and both LTC4 and LTD4 increase prostacyclin release by bovine aortic endothelial cells.49 Endothelial cells metabolize leukotrienes, which indicates that they have
receptors for them but do not synthesize leukotrienes.48 Although it might be argued that leukotrienes are local mediators of physiologic
responses and do not function as circulating hormones, this
may not be entirely true during pregnancy because of supplemental production
by the placenta. The lipoxins share some of the same biologic effects of the prostaglandins, HETEs, and
leukotrienes, but chemically they are distinct molecules. Lipoxin
A induces arteriolar dilation but does not affect microvascular
permeability, as does LTC4, or leukocyte adherence to venular endothelium, as does LTB4.1,2 Lipoxin A stimulates superoxide anion generation without provoking aggregation, causes
chemotaxis, and possesses spasmogenic activities, as
do the HETEs and leukotrienes. Lipoxin A elicits long-lasting contractions
of guinea pig lung strips, but, unlike LTC4, it does not stimulate contraction of the guinea pig ileum. Lipoxins A
and B inhibit human natural killer cell activity, whereas HETEs and leukotrienes
do not affect natural killer cell cytotoxicity. Furthermore, lipoxins
may serve as important intracellular mediators. Lipoxin A
is a potent activator of protein kinase C, even more potent than diacylglycerol
or arachidonic acid. Thus, lipoxins display patterns of physiologic
effects that are distinct from prostaglandins, thromboxanes, HETEs, or
leukotrienes. The importance of lipoxins in normal and abnormal
pregnancy is not known, but the fact that lipoxin A dilates arterioles
implies that they may serve to augment vasodilatation during normal
pregnancy. Uterine Blood Flow The effects of prostaglandins on uterine blood flow have been evaluated
in vivo in a number of species, including pregnant monkeys and pregnant
and nonpregnant chronically catheterized sheep. Radioactive microspheres
and uterine artery flow probes are the two techniques most commonly
used for evaluating uterine blood flow. When assessing the effects
on uterine blood flow of uterotonic agents, such as the prostaglandins, their
simultaneous effects on uterine contractility also must be determined. A
compound that stimulates uterine contractions also reduces
the amount of blood perfusing the uterus by myometrial compression of
the blood vessels. The prostaglandins that decrease uterine blood flow are PGE2, PGF2α, 6-keto-PGF1α, TXB2, and analogues of the prostaglandin endoperoxides PGG2 and PGH2.50,51,52,53,54 PGE2 and PGF2α are potent stimulators of uterine contractility. The effect of PGE2 on uterine blood flow is primarily due to its uterotonic action, whereas
PGF2α exerts vasoconstrictive actions in addition to its uterine contractile
effect. The prostaglandins that increase uterine blood flow are PGI2, PGD2, PGA2, PGE1, and 6-keto-PGE1. PGE2 is a vasodilator in nonpregnant sheep, provided it does not stimulate
uterine contractions.51,52,53,54,55,56,57,58 PGD2 and PGA2 stimulate uterine contractions, whereas PGI2 inhibits uterine activity. Placental Blood Flow IN VITRO STUDIES. Several in vitro studies have been published concerning the vasoactive
effects of thromboxane and prostaglandins on human placental vessels. These
studies demonstrate that thromboxane is one of the most potent
vasoconstrictors of the human placental vasculature.59,60,61,62,63 Because the active metabolite of thromboxane (TXA2) is extremely labile, it is necessary to use a stable analogue for experimental
studies. Most investigators use the thromboxane mimic U46619. In
vitro studies demonstrate that thromboxane is 10 to 1,000 times more
active as a vasoconstrictor than is PGE2, PGF2α, AII, 5-hydroxytryptamine (5-HT, or serotonin), norepinephrine (NE), or
bradykinin. With respect to the vasoconstrictive effects of prostaglandins in the human
placenta, there is agreement among all investigators that PGF2α and PGE2 are vasoconstrictors.60,61,62,63,64,65,66,67,68,69,70,71 PGD2 reportedly causes vasoconstriction when given alone,61,64 but attenuates vasoconstriction when given with AII.61 PGA1 causes vasoconstriction equivalent to PGF2α.71 The stable metabolite of thromboxane, TXB2, is without effect on the placental vasculature.71 In the human placenta, prostacyclin consistently is the most potent vasodilator.61,62,63,67,68,71 PGE1 was found to be a weak vasodilator in two studies,61,62 but a weak vasoconstrictor in another study.71 One of the stable metabolites of prostacyclin, 6-keto-PGE1, is a weak vasodilator in the human placenta.62 One of prostacyclin's other stable metabolites, 6-keto-PGF1α, is a weak vasodilator.25 There is little information available regarding the vascular effects of
leukotrienes in the placenta. When comparing the vasoconstrictive effect
of LTB4, LTC4, and LTD4 with that of thromboxane in the perfused human placental cotyledon,59 we found that the leukotrienes cause vasoconstriction, but only at high
doses. Their potency is considerably less than that of thromboxane. Based on in vitro studies using isolated human placental vascular strips
or isolated perfused human placental cotyledons, one can rank the vasoactive
potencies of thromboxane, prostaglandins, and other vasoactive
compounds as follows: 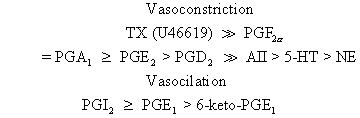
IN VIVO STUDIES. Several in vivo studies have been reported concerning the vasoactive effects
of prostaglandins in the placenta. The most commonly used animal
model is the chronically catheterized sheep, but other species have
been used, including nonhuman primates. Vasoconstrictors. There is agreement among investigators that fetal administration of PGE2 or PGF2α causes umbilical-placental vasoconstriction in the sheep.72,73,74,75,76 The thromboxane mimic U46619 is a potent vasoconstrictor in the ovine
fetal systemic circulation and in the umbilical-placental circulation.77,78 Furthermore, fetal infusion of thromboxane can result in respiratory acidosis
in the fetus, a further indication of its detrimental vasoconstrictive
effects on the fetoplacental vasculature. Thus, there is agreement
between in vitro and in vivo studies that thromboxane, PGE2, and PGF2α are vasoconstrictors in the fetoplacental vascular bed. Does Prostacyclin Vasodilate the Placental Vasculature In Vivo?In Vitro Versus In Vivo Results. With respect to vasodilatation in the fetoplacental vasculature, there
is controversy and disagreement between in vitro and in vivo studies. Prostacyclin
is a potent vasodilator,24,25,26 and it consistently relaxes human placental blood vessels when tested
in vitro, as discussed previously. However, when tested in vivo, infusion
of prostacyclin into the ovine fetus decreases fetoplacental blood
flow and results in an increase or no change in the calculated placental
vascular resistance.77,78,79 Furthermore, fetal infusion of prostacyclin into the systemic circulation
in conjunction with either thromboxane or AII reverses systemic and
renal vasoconstriction induced by thromboxane or AII but further decreases
fetoplacental blood flow.77 The in vivo placental vascular results obtained for prostacyclin in the
sheep are disturbing for at least two reasons. First, they contradict
the consistently potent vasodilatory effect of prostacyclin obtained
in human in vitro placental studies. Second, how can a compound that is
such a potent vasodilator in all other vascular beds be a vasoconstrictor
in the placenta, a vascular bed that must remain dilated if the
fetus is to grow and mature properly? The aberrant effects observed for prostacyclin in vivo can be explained
by Poiseuille's law as it applies to the fetal-placental circulation. The
placental vascular effects observed for prostacyclin in vivo (or
for other vasodilators) are passive changes caused by shunting of
blood away from the placenta to other vasodilated vascular beds more
proximal to the heart. We have previously described this concept in detail.80 The placental vascular actions are due to systemic alterations in blood
flow and perfusion pressure in nonplacental vascular beds rather than
to direct vasoconstrictive actions of prostacyclin on the placental vasculature. Indirect
evidence suggests that prostacyclin is important
for in vivo vasodilation in the human placenta. Doppler ultrasonographic
measurements of umbilical blood flow in pregnant women are correlated
positively with prostacyclin production in vitro by specimens obtained
from the umbilical arteries.81 Umbilical and Fetal Blood Flow The arachidonic acid metabolites are present in abundance82 and exert potent vasoactive effects in the fetus and on the umbilical
vessels. Thromboxane, PGE2, and PGF2α cause vasoconstriction, whereas prostacyclin causes vasodilatation of
the umbilical vessels.73,74,83,84,85,86 Thromboxane causes vasoconstriction of the ductus arteriosus and pulmonary
vasculature, whereas prostacyclin, PGE2, and PGE1 cause vasodilatation.87,88,89 Inhibitors of prostaglandin synthesis, such as indomethacin and sodium
salicylate (aspirin), result in vasoconstriction and closure of the ductus
arteriosus,90,91 demonstrating that prostaglandins are necessary to maintain patency of
the ductus arteriosus during fetal life. Thromboxane is a potent vasoconstrictor in the fetal systemic circulation, as
evidenced by in vivo studies in chronically catheterized sheep
fetuses. Thromboxane infusion results in a significant increase in mean
fetal aortic blood pressure, renal vasoconstriction, and fetal acidosis.77,78 Prostacyclin, however, vasodilates the ovine fetal systemic circulation, as
evidenced by its ability to significantly lower mean fetal aortic
blood pressure77,78,79 and its ability to antagonize the vasoconstrictive effects of thromboxane
and AII.77,78 Prostacyclin increases blood flow to the fetal kidneys,77 adrenal glands,92 and lungs.78,79 The leukotrienes are potent fetal pulmonary vasoconstrictors and may be
the compounds primarily responsible for maintaining the high pulmonary
vascular resistance during fetal life. In newborn lambs and piglets, LTD4 increases pulmonary and systemic vascular resistance,93,94 whereas blockage of the leukotriene receptors with FPL 57231 decreases
pulmonary vascular resistance, increases pulmonary blood flow, and decreases
pulmonary and systemic arterial pressures in late gestational
fetal lambs.95 Human newborns with pulmonary hypertension and persistent fetal circulation
have LTC4 and LTD4 present in their tracheal lavage; healthy newborns do not.96 Leukotriene inhibition with the receptor blocker FPL 57231 prevents and
reverses hypoxic pulmonary vasoconstriction in newborn lambs.97 |