LPD is a heterogeneous disorder that has been shown and investigated in
several primate species besides humans. As previously noted, the lack
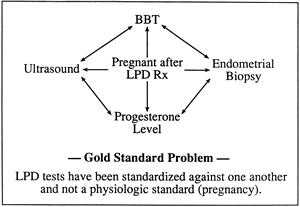
of a diagnostic gold standard has led to varying definitions and myriad
hypotheses as to the underlying defect within the menstrual cycle. There
is good evidence for the existence of several different causes for
LPD, one or more of which may exist in the same woman. In general, the
proposed pathophysiologic mechanisms may be divided into three categories
centered around the corpus luteum as the primary functional unit: preparation, production, and response (Fig. 7). The basic tenets of each are discussed. 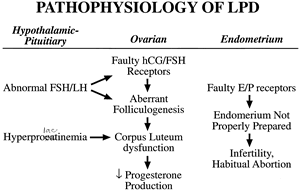 Fig. 7. Luteal phase deficiency is a heterogeneous disease with a multifactorial
cause. It can result from abnormalities at the level of the hypothalamus/pituitary, the
ovary, or the endometrium. Fig. 7. Luteal phase deficiency is a heterogeneous disease with a multifactorial
cause. It can result from abnormalities at the level of the hypothalamus/pituitary, the
ovary, or the endometrium.
|
Preparation: Inadequate Folliculogenesis Because the corpus luteum develops from its predecessor, the dominant follicle, it
is logical to assume that any defect in the formation or function
of the latter could lead to LPD. Early investigation in nonhuman
primates by DiZerega and Hodgen39 and Stouffer and Hodgen40 showed that diminished FSH secretion in the early follicular phase induced
by administration of porcine follicular fluid rich in inhibin led
to inadequate luteal progesterone secretion. They showed that inadequate
follicular phase levels of FSH and estradiol ultimately led to incomplete
differentiation of luteal cells that were less sensitive to LH
and hCG stimulation. Wilks and colleagues41 previously described a group of rhesus macaques who had spontaneous LPD (decreased
progesterone levels) and decreased follicular phase FSH levels. Human
studies also showed lower follicular FSH levels or diminished
FSH-to-LH ratios in women with LPD diagnosed by serum progesterone
or histologic criteria.42,43,44,45,46 The exact cause for diminished FSH secretion or effect in spontaneous
menstrual cycles with LPD is unknown, but there has been speculation that
this finding is due to inhibin or inhibin-like substances that either
suppress pituitary FSH secretion or alter its bioactivity. An investigation
by Molskness and colleagues47 showed diminished bioactive FSH and estradiol secretion, increased bioactive
LH secretion, and lengthening of the follicular phase in rhesus
monkeys treated with human inhibin A early in their menstrual cycles. These
animals experienced decreased mean luteal progesterone levels despite
normal luteal phase length and normal levels and patterns of luteal
LH secretion compared with controls. A similar association between
a prolonged follicular phase and subsequent LPD has been noted in spontaneous
human cycles.11,48 Despite this compelling evidence that abnormalities of follicular phase
FSH lead to LPD, other investigations using more frequent sampling techniques
in spontaneous cycles with out-of-phase biopsy specimens and
diminished integrated luteal progesterone levels showed no differences
in follicular phase FSH (immuno- and bioactive) and estradiol levels
compared with controls.11,49 To confuse the situation further, and in contrast to the animal studies
previously noted, lower levels of inhibin were detected in the early
follicular and luteal phases of cycles in women with LPD.49,50 Altogether, we currently believe that abnormalities in folliculogenesis
with or without FSH deficiencies can cause LPD, but this mechanism is
not the common pathogenesis for LPD as found in most women. Although some investigators have implicated follicular phase FSH as a crucial
factor in later corpus luteum function, others have focused on
follicular phase LH—specifically, its pattern of pulsatile secretion
early in the menstrual cycle. Soules and coworkers51 first reported abnormal pulsatile patterns of LH secretion in four fertile
women with histologically diagnosed LPD. They showed an increased
early follicular LH pulse frequency compared with control women. In a
larger and more definitive study,52 these investigators showed that women with LPD maintain a fixed pattern
of increased pulsatile secretion throughout the follicular phase, in
contrast to normal women, who experience an acceleration in pulse frequency
with the approach of ovulation. This pattern emerged despite similar
follicular phase peak and integrated estradiol levels and no change
in follicular phase mean LH and FSH (immuno- and bio-levels).49 They speculated that the increased LH pulse frequency may result from
lower serum progesterone levels in the preceding luteal phase because
progesterone has been shown to slow LH secretion by the pituitary.51 Soules and coworkers53 tested this latter hypothesis in a study of six normal women with LPD
induced by administration of a GnRH antagonist in the midluteal phase. Analysis
of LH pulses in the early follicular phase of the ensuing menstrual
cycle showed no increased frequency over baseline, however, suggesting
that the intrinsic defect lay within the neuroendocrine control
of the GnRH pulse generator and not in the antecedent corpus luteum. These
findings were confirmed by a similar pulse analysis study by Suh
and Betz.54 The immediate sequela of an increased LH pulse frequency in the first half
of the menstrual cycle seems to be diminished levels of bioactive
LH in the luteal phase, with concomitant decreases in progesterone pulse
amplitude and mean serum progesterone levels (Fig. 8).49,52 These observations in spontaneous menstrual cycles confirmed an earlier study in which luteal bioactive LH was diminished after a supraphysiologic
gonadotropin pulse frequency was induced in the follicular phase
in normal women.55 In a study of a group of young women with exercise-related and diet-related
LPD, analysis of early follicular LH pulses showed slower frequencies
compared with normal women.56 This seeming contradiction may serve only to show how perturbation of
the gonadotropin pulse generator in one direction or another during the
follicular phase can alter subsequent corpus luteum function. 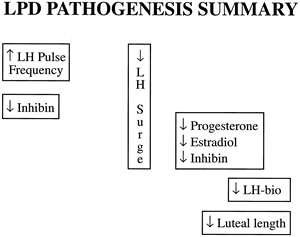 Fig. 8. Our research group identified a series of reproductive hormone abnormalities
that are associated with luteal phase deficiency. Based on our studies, we
describe the pathophysiology of luteal phase deficiency as
follows. There is a supraphysiologic luteinizing hormone pulse frequency
throughout the luteal phase, which is followed by a luteinizing hormone
surge of decreased magnitude. After ovulation, the corpus luteum
secretes lesser amounts of all hormones, including progesterone, estradiol, and
inhibin. Luteinizing hormone as atrophic hormone of the corpus
luteum is decreased in its biopotency in the mid to late luteal phases. These
events lead to a premature luteolysis and a subtle decrease
in luteal phase duration and subsequently menstrual cycle length. Fig. 8. Our research group identified a series of reproductive hormone abnormalities
that are associated with luteal phase deficiency. Based on our studies, we
describe the pathophysiology of luteal phase deficiency as
follows. There is a supraphysiologic luteinizing hormone pulse frequency
throughout the luteal phase, which is followed by a luteinizing hormone
surge of decreased magnitude. After ovulation, the corpus luteum
secretes lesser amounts of all hormones, including progesterone, estradiol, and
inhibin. Luteinizing hormone as atrophic hormone of the corpus
luteum is decreased in its biopotency in the mid to late luteal phases. These
events lead to a premature luteolysis and a subtle decrease
in luteal phase duration and subsequently menstrual cycle length.
|
Whether inhibin is part of the pathophysiology of LPD is uncertain. As
previously noted, early and mid follicular levels of inhibin have been
noted to be diminished in LPD cycles despite normal levels of FSH and
estradiol.49,50 Soules and coworkers49 speculated that lower levels of inhibin may reflect a differential insensitivity
to FSH that does not affect estradiol secretion. The difficulty
in proving such a hypothesis was the significant amount of overlap
between inhibin levels in normal and LPD women. In addition, the relative
lack of specificity of the inhibin assay available at the time of
that analysis did not allow accurate quantification or identification
of the active dimeric forms of inhibin (A and B). A more recent study
by Molskness and colleagues,47 in which supraphysiologic levels of inhibin A were given to monkeys to
induce LPD, further clouds the issue of inhibin. It would be interesting
to know whether there are any changes in circulating levels of inhibin
A or B during the menstrual cycles of women with spontaneous LPD. Studies of ovulation induction in which ovulation was triggered using small
amounts of hCG or LH by Jones and Madrigal-Castro57 and Van de Wiele and coworkers58 first raised the possibility of mid cycle LH surge inadequacy as a potential
mechanism for LPD. These investigators showed that an inadequate
surge duration led to aberrant luteal function. Later investigations
using daily blood sampling confirmed that LPD cycles were characterized
by attenuated integrated LH levels around the time of the mid cycle
surge (Fig. 9).49,54 The authors hypothesized that the more frequent but stunted LH pulses
throughout the follicular phase may down-regulate LH secretion at mid
cycle, leading to a blunted LH surge.54 At present, there are no studies to prove or disprove this theory. These
findings in LPD cycles are difficult to explain, considering a lack
of correlation between integrated LH surge levels and progesterone production
in normal menstrual cycles.59 Taken together, one might hypothesize that there is a minimal threshold
for an LH surge that ensures, but does not control, the amount of progesterone
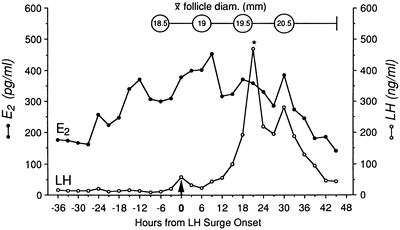
subsequently secreted. 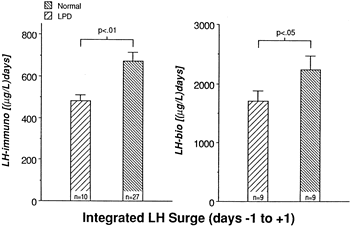 Fig. 9. Integrated luteinizing hormone (LH) surge, days −1 to +1. Daily
blood samples were obtained in normal women and women with luteal
phase deficiency across the menstrual cycle. The peak LH level at mid
cycle was identified as the day of the LH surge (day 0). The elevated
daily LH levels on either side of day 0, which were part of the LH
surge, were combined (integrated) and compared between the two groups. The
integrated LH surge levels for women with luteal phase deficiency
were significantly lower than normal when LH was assayed in a standard
radioimmunoassay and a Leydig cell bioassay. Fig. 9. Integrated luteinizing hormone (LH) surge, days −1 to +1. Daily
blood samples were obtained in normal women and women with luteal
phase deficiency across the menstrual cycle. The peak LH level at mid
cycle was identified as the day of the LH surge (day 0). The elevated
daily LH levels on either side of day 0, which were part of the LH
surge, were combined (integrated) and compared between the two groups. The
integrated LH surge levels for women with luteal phase deficiency
were significantly lower than normal when LH was assayed in a standard
radioimmunoassay and a Leydig cell bioassay.
|
Other investigations of LPD cycles have focused on the development and
function of the preovulatory (dominant) follicle. Check and colleagues60 evaluated women with histologically proven LPD sonographically and found
that they ovulated more frequently from relatively small dominant follicles (<17 mm
mean diameter). This finding was confirmed by
Ying and associates,61 who noted that 39% of LPD cycles were characterized by small dominant
follicles compared with those of normal women at their institution. They
also found that this percentage decreased to 6% in histologically
corrected LPD cycles. These findings were later refuted, however, by
other investigations of spontaneous LPD cycles during which
daily ultrasound monitoring of follicles was performed.11,49 Production: Inadequate Progesterone The endocrine function of the corpus luteum, as is the case for other endocrine
organs, depends on the interplay of multiple factors: (1) Sufficient
precursor molecules must be available for eventual conversion
to the active hormone; (2) tropic stimuli must be present in the proper
quantity and quality to promote hormone synthesis; (3) the target cells
of the tropic stimuli must have the requisite mechanism for receiving
and converting that message for intracellular response; (4) the actual
cellular production of hormone must be adequate; and (5) the activity
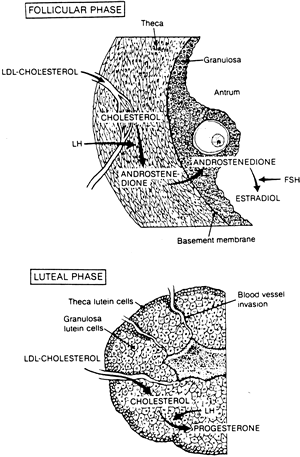
of the hormone secreted must be sufficient. The precursor for biosynthesis of progesterone is plasma cholesterol transported
as LDL. According to the model presented by Carr and colleagues,26 the poorly vascularized granulosa cells in the follicular phase normally
are isolated from plasma LDL cholesterol (see Fig. 3). After ovulation, as the microvascular network in the thecal layers invades
the luteinized granulosa cells, local LDL concentrations approach
plasma levels, providing the substrate for progesterone production. In
support of this substrate theory, observers of corpus luteum anatomy
have found maximal capillary enlargement or dilaton to have occurred
by luteal day 7 or 8, the time of maximal progesterone secretion.62,63 Conceivably, inadequate vascularization could undermine the process of
progesterone production, as has been noted histologically in several
case studies.64,65 Alternatively, short supply or abnormal forms of LDL cholesterol could
lead to inadequate corpus luteum output. Illingworth and colleagues66 reported that women with abetalipoproteinemia, a syndrome marked by extremely
low amounts of circulating LDL, have very low serum levels of
progesterone (<2 ng/mL) despite otherwise normal menstrual cycle
parameters. Serum levels of LDL decrease during the luteal phase in
normal menstrual cycles67; however, no difference in luteal phase serum lipoprotein levels between
women with normal and LPD cycles was found in an investigation by Hansen
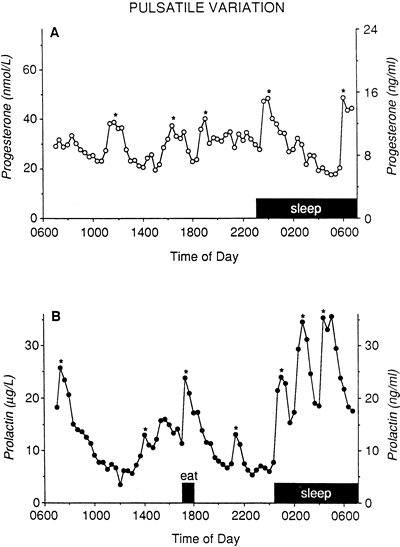
and colleagues.68 Progesterone is secreted in a pulsatile fashion by the corpus luteum in
normal and LPD women in response to LH secretion by the pituitary (Fig. 10).31,52,59 Intuitively, disruption of LH secretion could lead secondarily to deficiencies
in progesterone production by the corpus luteum. Soules and coworkers52 studied luteal phase gonadotropin secretion during LPD cycles and showed
that there was no significant difference in LH pulse frequency or amplitude
compared with controls. The women studied experienced normal
slowing of pulse frequency and an increase in pulse amplitude during the
transition from follicular to luteal phases. This slowing of the LH
secretion pattern previously was shown to be controlled by progesterone
feedback on the GnRH pulse generator, mediated via endogenous opioid
peptides (Fig. 11).51 Despite apparently normal patterns of luteal phase LH secretion, bioactive
levels of LH were decreased significantly 6 to 11 days after a mid
cycle surge, suggesting that there is a qualitative defect in the stimulus
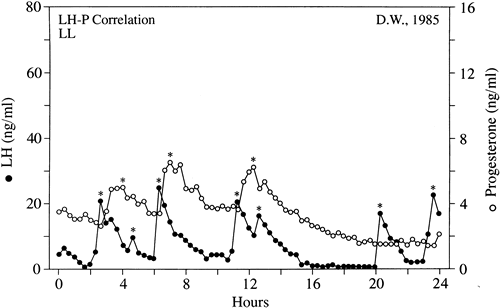
to luteal cells.49 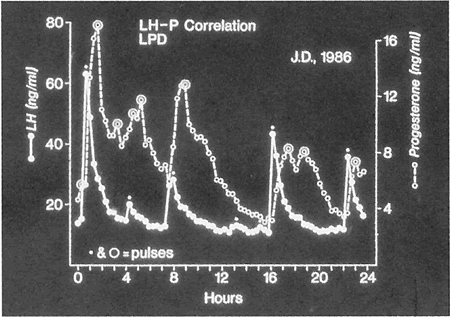 Fig. 10. Progesterone secretion in luteal phase deficiency. Blood samples drawn
every 20 minutes for 24 hours in a woman with low integrated progesterone
secretion over the duration of her luteal phase (LPD) were assayed
for luteinizing hormone (LH) and progesterone (P). The LH changes marked
with an asterisk are true secretory pulses, as are the progesterone
data points that are circled. The synchrony of LH and progesterone secretion
was maintained in women with luteal phase deficiency in the same
manner as occurs in normal women. Fig. 10. Progesterone secretion in luteal phase deficiency. Blood samples drawn
every 20 minutes for 24 hours in a woman with low integrated progesterone
secretion over the duration of her luteal phase (LPD) were assayed
for luteinizing hormone (LH) and progesterone (P). The LH changes marked
with an asterisk are true secretory pulses, as are the progesterone
data points that are circled. The synchrony of LH and progesterone secretion
was maintained in women with luteal phase deficiency in the same
manner as occurs in normal women.
|
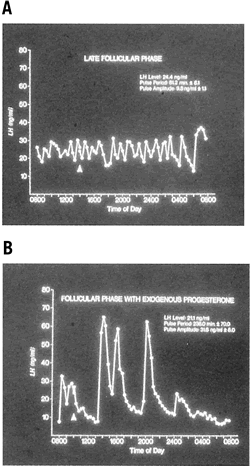 Fig. 11. A. Progesterone feedback on the gonadotropin-releasing hormone pulse generator. The
graphs on the left and right are 24-hour secretory patterns
of luteinizing hormone (LH) in a normal woman. Samples were obtained
in the mid follicular phase of two corrective cycles. The graph on the
left depicts a relatively rapid LH pulse pattern with an even, relatively
low pulse amplitude. The LH secretory pattern on the right occurred
in the same woman in a subsequent cycle after she had received 7 days
of exogenous progesterone. The exogenous progesterone converted her
LH secretory pattern to a pattern that was indistinguishable from the
normal luteal phase pattern (lower pulse frequency and higher pulse amplitude). B. Progesterone feedback on the gonadotropin-releasing hormone pulse generator. The
graphs on the left and right are 24-hour secretory patterns
of LH in a normal woman. Samples were obtained in the mid follicular
phase of two corrective cycles. The graph on the left depicts a relatively
rapid LH pulse pattern with an even, relatively low pulse amplitude. The
LH secretory pattern on the right occurred in the same woman in
a subsequent cycle after she had received 7 days of exogenous progesterone. The
exogenous progesterone converted her LH secretory pattern
to a pattern that was indistinguishable from the normal luteal phase pattern (lower
pulse frequency and higher pulse amplitude). Fig. 11. A. Progesterone feedback on the gonadotropin-releasing hormone pulse generator. The
graphs on the left and right are 24-hour secretory patterns
of luteinizing hormone (LH) in a normal woman. Samples were obtained
in the mid follicular phase of two corrective cycles. The graph on the
left depicts a relatively rapid LH pulse pattern with an even, relatively
low pulse amplitude. The LH secretory pattern on the right occurred
in the same woman in a subsequent cycle after she had received 7 days
of exogenous progesterone. The exogenous progesterone converted her
LH secretory pattern to a pattern that was indistinguishable from the
normal luteal phase pattern (lower pulse frequency and higher pulse amplitude). B. Progesterone feedback on the gonadotropin-releasing hormone pulse generator. The
graphs on the left and right are 24-hour secretory patterns
of LH in a normal woman. Samples were obtained in the mid follicular
phase of two corrective cycles. The graph on the left depicts a relatively
rapid LH pulse pattern with an even, relatively low pulse amplitude. The
LH secretory pattern on the right occurred in the same woman in
a subsequent cycle after she had received 7 days of exogenous progesterone. The
exogenous progesterone converted her LH secretory pattern
to a pattern that was indistinguishable from the normal luteal phase pattern (lower
pulse frequency and higher pulse amplitude).
|
In a similar fashion in conceptive cycles, continuation of pregnancy depends
on tropic support of the corpus luteum by hCG elaborated by the
early trophoblast. Apparently the corpus luteum becomes increasingly less
sensitive to LH stimulation with a preferential response to hCG support
as ovulation becomes more remote. Without adequate hCG, luteolysis
takes place, with ensuing pregnancy failure. Conceivably, alterations
in the hCG molecule affecting its bioactivity or in the pattern of
its secretion could disturb this luteal rescue function. This hypothesis
is theoretical at present but offers a provocative target for future
research. The aforementioned two-cell compartmentalization of the corpus luteum has
led to studies of LPD based on hypothetical failure of specific cell
types. Hinney and colleagues28 investigated LH pulsatility in 38 women with LPD diagnosed by two consecutive
cycles marked by diminished mid luteal progesterone levels. They
described three distinct patterns of luteal phase failure. Group 1 consisted
of 16 women (42%) who had no LH secretory episodes and
had significantly lower basal LH levels compared with 14 control subjects. Group 2 consisted
of 13 women (34%) who had normal LH secretory
episodes despite low estradiol and progesterone concentrations
that did not respond to the LH pulses. Group 3 consisted of nine women (24%) who
had normal LH pulses with an adequate progesterone
response but inadequate basal secretion of progesterone. All immunoreactive
LH pulses also were found to be bioactive. Group 1 subjects were
designated as having a hypothalamic cause of LPD (inadequate stimulation). Group 2 women, because of their lack of response to LH pulses, were
categorized as having a small cell defect. Group 3 women, with low
basal levels of progesterone and appropriate responses to LH episodes, were
categorized as having a large cell defect. These findings led to
intriguing implications for the management of patients with LPD. Patients
found to have an increase in serum progesterone levels in response
to LH stimulation (large cell defect) may be treated adequately with
exogenous hCG; patients with inadequate LH response (small cell defect) may
be treated preferentially with progesterone supplementation. Further
investigation is needed to verify these findings and to characterize
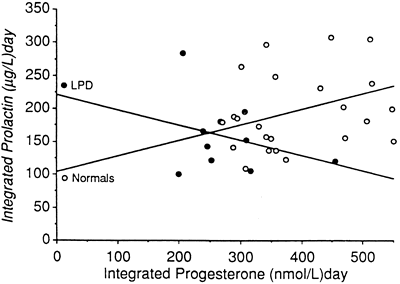
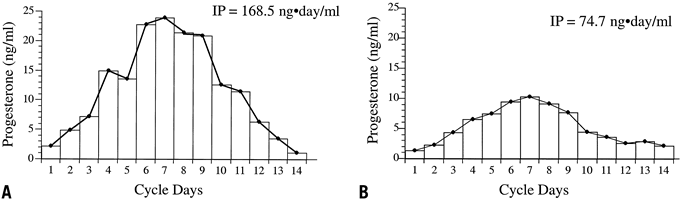
the specific cellular defects within these subtypes. Further analysis of LPD has focused on the quantity and quality of progesterone
produced by the corpus luteum. The difficulty in assessing the
former is the wide fluctuation in serum progesterone levels over a 24-hour
period. One study using frequent blood sampling in normal women
during the mid and late luteal phases found the mean percentage variation
in progesterone over 24 hours to be 99% and 138% (Fig. 12).59 No identifiable circadian pattern of secretion was established. This level
of variation affects the interpretation of single or multiple values
in the assessment of luteal adequacy. By increasing the number of
data points, however, one may limit the amount of variance and obtain
a more accurate estimate of the true mean. This principle, previously
referred to as regression toward the mean, allows accurate comparison of women and groups in terms of their corpus
luteum secretory capacity using integrated daily luteal phase progesterone
levels. Numerous studies that have undertaken such measurements
have confirmed that a subset of women exist with decreased quantities
of progesterone compared with controls. In some cases, the typical bell-shaped
curve of progesterone levels is attenuated, and in others, progesterone
peaks early, with significant shortening of the overall luteal
phase duration. Intuitively, in cases in which histologic LPD exists
despite adequate immunoreactive levels of progesterone in the serum, there
may be abnormalities of the bioactive fraction of progesterone. A
study by Minassian and Wu70 specifically addressing this issue could not show a difference, however, in
the amount of free and protein-bound progesterone between normal
and histologic LPD cycles. 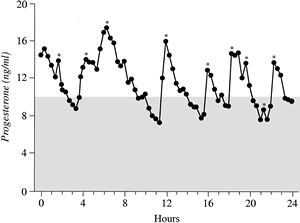 Fig. 12. Progesterone secretion pattern. Blood samples taken every 20 minutes over 24 hours
from a normal woman were analyzed for progesterone. Her consecutive
serum progesterone levels are graphed with secretory events (pulses) marked
with an asterisk. By careful examination, it can be seen
that her progesterone levels vary from 8 to 18 ng/mL over 24 hours. About
a third of the time, progesterone levels were less than 10 ng/mL, which
has been suggested by some authors as a crucial level for the
diagnosis of luteal phase deficiency. This graph shows that single blood
samples can be deceptive, depending on where they were obtained during
the progesterone secretory profile. Fig. 12. Progesterone secretion pattern. Blood samples taken every 20 minutes over 24 hours
from a normal woman were analyzed for progesterone. Her consecutive
serum progesterone levels are graphed with secretory events (pulses) marked
with an asterisk. By careful examination, it can be seen
that her progesterone levels vary from 8 to 18 ng/mL over 24 hours. About
a third of the time, progesterone levels were less than 10 ng/mL, which
has been suggested by some authors as a crucial level for the
diagnosis of luteal phase deficiency. This graph shows that single blood
samples can be deceptive, depending on where they were obtained during
the progesterone secretory profile.
|
Response: Effects of Progesterone on the Endometrium An area of continued interest and controversy in the investigation of LPD
pathophysiologic mechanisms is the endometrial response to ovarian
steroids. Normally, over the course of the menstrual cycle, the endometrium
undergoes dynamic changes in receptivity in response to hormone
signals from the ovaries. In general, estradiol and progesterone receptor
content increases throughout the proliferative phase, with peak levels
in the immediate preovulatory period.71,72 During the secretory phase, under the influence of progesterone, estradiol
receptor content declines in all cell types, and progesterone receptors
are reduced in glandular epithelium, while remaining constant in
stromal and myometrial cells.71 Retention of progesterone receptors in the latter two categories is thought
to aid in the maintenance of pregnancy by inhibiting myometrial
contractility during gestation. Given these complex and essential functions
of progesterone, dependent on the adequacy of its receptor, it has
been compelling to address this area as a potential cause of LPD. The first description of a patient with an abnormal progesterone receptor
concentration in endometrium came from Keller and coworkers in 1979.73 In this case report, normal serum levels of estradiol, progesterone, and
FSH were documented in the face of multiple abnormal luteal phase endometrial
biopsy specimens. Subsequent progesterone supplementation failed
to correct the histologic finding of a poorly developed pseudodecidual
endometrial reaction. Eventual progesterone binding studies on
a sample of the patient’s endometrium revealed normal receptor
affinity but a concentration of progesterone receptors only half that
of endometrium from control subjects. This case report was a stimulus
for investigation and elaboration of steroid receptor changes in the endometrium
in women with histologic LPD (Table 1). TABLE 1. Summary of Estrogen and Progesterone Receptor Concentrations in
Endometrial Biopsy Specimens from Women with Luteal Phase Deficiency
| |
No. Subjects | Progesterone Receptors | Estrogen Receptors |
|
Study |
LPD |
Normal |
Cytosolic |
Nuclear |
Cytosolic |
Nuclear |
|
Gautray (1981)74 |
88 |
79 |
Decreased |
Decreased |
Decreased |
Decreased |
|
Gravanis (1984)75 |
10 |
7 |
NS |
Increased |
— |
— |
|
Laatikainen (1983)76 |
14 |
19 |
Decreased |
— |
— |
— |
|
Levy (1980)77 |
18 |
16 |
NS |
NS |
NS |
NS |
|
McRae (1984)78 |
14 |
30 |
NS |
— |
NS |
— |
|
Saracoglu (1985)79 |
20 |
40 |
Increased |
— |
NS |
— |
|
Spirtos (1985)80 |
10 |
14 |
Decreased |
|
|
|
LPD, luteal phase deficiency; NS, not significant.
McNeely MJ, Soules MR: The diagnosis of luteal phase deficiency: A critical
review. Fertil Steril 50:1, 1988.
Table 1 shows there is little consensus on this issue. All of these studies specifically
examined coincidental secretory phase parameters (i.e., serum
estradiol and progesterone levels) rather than proliferative phase
steroids, which have a more important role in induction of endometrial
receptors. As others have suggested,81,82 the absolute number or density of steroid receptors may not be the problem
as much as the ratio of estradiol to progesterone receptors in the
nucleus and cytosol. In one investigation by Abd-El-Maeboud and coworkers,83 quantification of estradiol and progesterone receptors using a sensitive
monoclonal antibody technique in women with histologic LPD revealed
significantly lower total progesterone-to-total estradiol receptor ratios
compared with other infertile controls. By comparing receptor ratios
rather than absolute concentrations between groups, the authors minimized
the variability in the latter induced by differing ovarian stimulation
regimens. Further investigation of complete menstrual cycles
with unbiased morphometric and quantitative evaluation of endometrial
steroid receptors is necessary before it can be concluded that there is
a unifying endometrial cause for clinical LPD. From another perspective of a potential endometrial cause, the endometrial
response need not be devoid or attenuated but simply delayed. A phase
shift could result in suboptimal preparation of the endometrium for
attachment and nidation of the blastocyst at the time it enters the
uterus. This type of in utero embryonic asynchrony has been well documented
in myriad animal species.84 The existence of a window of implantation in the human uterus was established
by Hertig and colleagues,85 who examined hysterectomy specimens from women in the luteal phase of
their cycles. They discovered that all embryos (n = 8) from hysterectomies
performed before day 20 were free-floating, but all embryos (n = 26) from
hysterectomies performed after day 20 had implanted. Likewise, several
other investigations of the time of maximal uterine
receptivity86,87 have placed the window of implantation between postovulatory days 6 and 10 in
humans. If the coordinated timing of tubal embryo transport and
endometrial receptivity were disrupted, the putative window may be open
too late or too briefly for adequate implantation, leading to failed
pregnancy or early pregnancy wastage. For this reason, biopsy of the
endometrium in the mid to late secretory phase emerged as an attractive
bioassay of luteal phase sufficiency. The validity of endometrial
biopsy for the diagnosis of LPD and the optimal time to obtain a biopsy
specimen remain questionable (see later). Histologically the mechanism of failure may not depend on steroid hormones
and their receptors at all, but rather on the failure of expression
of necessary cell surface adhesion molecules (integrins). Lessey and
colleagues88 described two types of integrin defects during the mid luteal phases of
women with previously unexplained infertility: type I, patients with
out-of-phase endometrial biopsy specimens (i.e., LPD) lacking β3 integrin, and
type II, patients with in-phase biopsy specimens but
still lacking β3 integrin. Even when type I patients were treated
adequately, almost one third (5 of 16) failed to express the β3 integrin. Despite
correction of a steroid deficiency in the endometrium, a
group of women exists who have an endometrium that may be incapable
of expressing certain molecules integral to normal implantation
and pregnancy maintenance. To date, failed integrin expression in endometrial
biopsy specimens has proved to be useful as a diagnostic tool
for failed implantation; however, no known, reliable corrective measures
exist.89 |