Dynamic Mutations As noted above, there are exceptions to Mendelian inheritance. One assumption
of classic Mendelian genetics is that mutations are stably transmitted (i.e., they are passed unchanged from parent to offspring). The more severe
phenotype and earlier onset of disease in each succeeding generation in
families with fragile X syndrome (FX) or myotonic dystrophy (DM) were
thus quite puzzling. This so-called anticipation was even considered
to be the result of ascertainment bias until it was discovered in 1991 that
the genes for spinobulbar muscular atrophy and FX contain unstable
trinucleotide repeat sequences that become larger on each germline
transmission. DM and a number of AD spinocerebellar ataxias (SCAs) are
also caused by unstable or dynamic mutations. There currently are 13 autosomal
and two XL inherited diseases known to be caused by triplet
repeat expansions (Table 1), as well as a number of cytogenetic abnormalities or fragile sites that
are not associated with disease. It is still not clear why the expansions
occur, but slippage of the DNA polymerase enzyme during DNA replication
likely plays a role. The trinucleotide repeat sequences are CGG, CAG, CTG, GAA, or
CGG, and their normal size ranges and disease-causing
expansions are given (see Table 1). The triplet repeat may be of an intermediate (premutation) size that
does not itself cause disease but is prone to expand to a full mutation
during transmission to the next generation, leading to disease in the
offspring. The likelihood and degree of expansion may depend on the
sex of the transmitting parent for many of the disorders: expansions
are more likely during maternal transmission in FX, DM, and FA and during
paternal transmission in Huntington's disease and most SCAs. The
triplet repeats are all in the 5'UTR, 3'UTR, or coding
regions of the respective genes except in the case of the AR Friedreich's
ataxia and SCA8. The repeats have different, incompletely understood
effects on the function of the mutated proteins. In FX, the expanded
repeat in the 5'UTR leads to decreased transcription and
absent protein; in the SCAs, where the mutation is in the coding region
of the gene, the protein becomes deleterious to the cell, likely because
of an altered conformation that renders it less degradable.4,5 TABLE 1. Disorders Caused by Triplet Repeat Amplifications
|
Disorder | Inheritance | Gene | WT Size | Mutant Size | Parental Bias |
Fragile X syndrome | XL | FMR1 in Xq27.3 (FRAXA) | (CGG) 6---52 | (CGG) 60---200 (premutation)(CGG) 230---1000 (full) | Maternal |
Fragile XE mental retardation | XL | FMR2 in Xq28 (FRAXE) | (GCC) 7---35 | (GCC) 130---150 (premutation)(GCC) 230---750 (full) | Not determined |
Friedreich's ataxia | AR | X25 in 9q13---21.1 | (GAA) 6---34 | (GAA) 80 (premutation) (GAA) 112---1700 (full) | Maternal |
Myotonic dystrophy | AD | DMPK in 19q13 | (CTG) 5---37 | (CTG) 50---3000 | Maternal |
Spinobulbar muscular atrophy (Kennedy's disease) | AD | AR in Xq13---21 | (CAG) 11---33 | (CAG) 38---66 | Not determined |
Huntington's disease | AD | IT15 in 4p16.3 | (CAG) 6---39 | (CAG) 36---121 | Paternal |
Dentatorubropallidoluysian atrophy | AD | DRPLA (B37) in 12p31.31 | (CAG) 6---35 | (CAG) 51---88 | Paternal |
Spinocerebellar ataxia type 1 | AD | SCA1 in 6p23 | (CAG) 6---44 | (CAG) 39---82 | Paternal |
Spinocerebellar ataxia type 2 | AD | SCA2 in 12q24.1 | (CAG) 15---32 | (CAG) 36---63 | Paternal |
Spinocerebellar ataxia type 3 Machado-Joseph disease | AD | SCA3 (MJD1) in 14q32.1 | (CAG) 12---41 | (CAG) 62---84 | Paternal |
Spinocerebellar ataxia type 6 Episodic ataxia type 2 | AD | CACNA1A in 19p13 | (CAG) 14–18(CAG) 21---27 (SCA6) | (CAG) 21---23 (EA2) | Not determined |
Spinocerebellar ataxia type 7 | AD | SCA7 in 3p12---13 | (CAG) 5---34 | (CAG) 37---306 | Paternal |
Spinocerebellar ataxia type 8 | AD | SCA8 in 13q21 | (CTA/CTG) 16---91 | (CTA/CTG) 110---130 | Maternal | Two entities in this group of disorders are particularly noteworthy: FX
syndrome and myotonic dystrophy. FX syndrome, one of the most common
forms of mental retardation in males (incidence 1/4000), is characterized
by moderate-to-severe mental retardation with hyperactivity, autistic
features, and facial dysmorphism (e.g., broad jaw, large ears). The phenotype is caused by an expanded CGG repeat (200 to 1000 repeats) in the 5'UTR of the FX mental retardation 1 (FMR1) gene on chromosome Xq27.3. A repeat size above 200 leads to the full
syndrome and is associated with a folate-sensitive fragile site on chromosome
metaphase spreads. This characteristic led to the name of the
syndrome and was used to confirm the diagnosis before the availability
of more sensitive molecular testing (repeat-size detection by Southern
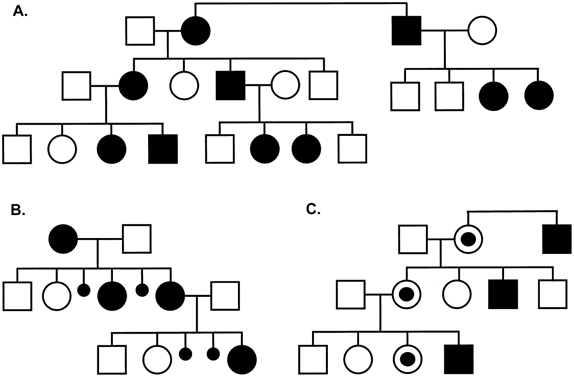
analysis). Figure 6 illustrates the XL inheritance and additional features that are typical
of FX pedigrees. Approximately 30% of female carriers of full mutations
have mild-to-severe mental retardation. There are also unaffected transmitting
males in FX families who can transmit a premutation to their
phenotypically normal daughters, who themselves can have affected sons
with full mutations. This dependence of the expression of the FX phenotype
on the position in the pedigree is called the Sherman paradox
and was based on the observation that the likelihood for mental impairment
was higher in the offspring of intellectually normal daughters of
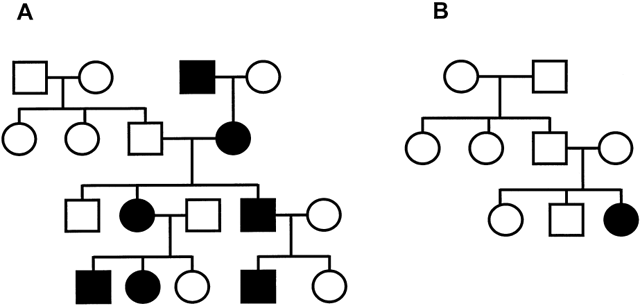
transmitting males than in the transmitting males' siblings. 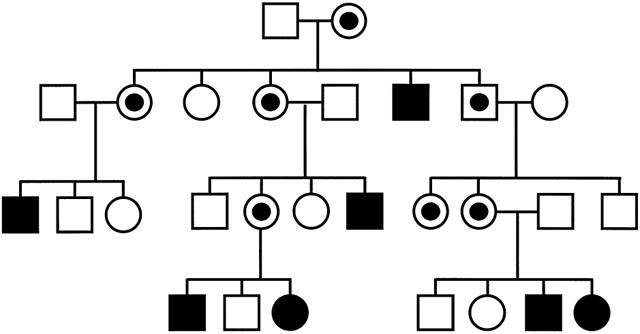 Fig. 6. Typical pedigree of fragile X syndrome. Note the presence of a transmitting
male and anticipation with more affected individuals in later generations. Fig. 6. Typical pedigree of fragile X syndrome. Note the presence of a transmitting
male and anticipation with more affected individuals in later generations.
|
The molecular diagnostic test for the CGG repeat expansion in FX is highly
reliable and should be offered to all patients with a family history
of FX or mental retardation of unknown cause. Prenatal diagnosis can
detect affected males in asymptomatic full or premutation carriers. Female
fetuses with a full mutation allele have up to a 50% chance of
having mental impairment. Female carriers of premutations are at increased
risk of premature ovarian failure when they inherit the mutation
from their father.4,5,6 Myotonic dystrophy is one of the most common inherited muscular disorders
and is characterized by myotonia, muscle weakness and atrophy, cardiac
conduction system anomalies, frontal balding, cataracts, endocrine
abnormalities, and mild mental impairment. It is caused by a trinucleotide
CTG repeat expansion in the 3'UTR of the myotonic dystrophy
protein kinase gene. There is clear anticipation, with earlier onset
and more severe disease in later generations. When the mutation is transmitted
through the maternal germline, massive CTG expansions (more
than 1000 repeats) can occur, leading to a congenital form of the disease
that causes severe hypotonia with facial diplegia, neonatal respiratory
insufficiency, and mental retardation. Women carrying an affected
fetus are at risk of polyhydramnios and preterm labor. Because of the
muscle weakness and cardiac conduction system anomalies, tocolysis with
magnesium sulfate can cause life-threatening respiratory depression
and arrhythmias.4,5,7 Genomic Imprinting A central assumption of Mendel's laws of inheritance is that genes
originating from maternal and paternal genomes are equally expressed
in the offspring. In some disorders, however, such as the Beckwith-Wiedemann
syndrome, the Prader-Willi syndrome (PWS), and the Angelman syndrome (AS), the
sex of the transmitting parent plays a role in the expression
of the phenotype in his or her affected children. This led to
the discovery that for some genes, only the allele inherited from a particular
parent is expressed in the offspring. A gene that is expressed
only from the paternally inherited chromosome is maternally imprinted (the
maternal allele is inactivated); a gene that is expressed only
from the maternally inherited chromosome is paternally imprinted (the
paternal allele is inactivated). When such a gene is mutated, only individuals
in whom the mutated allele is expressed will have the disease, whereas
individuals whose mutated copy is imprinted will be healthy. Figure 7 shows a characteristic pedigree for a maternally imprinted disorder: whenever
the mutation passes through the maternal germline, the offspring
are nonmanifesting carriers. Her sons will have affected children, whereas
her daughters will again silently transmit the mutation to the
next generation. Overall, there should be an equal number of affected
individuals and nonmanifesting carriers in each generation. Such genes, therefore, must
carry a reversible tag or imprinting mark capable of
switching when it passes through the germline of either sex. Once this
mark has switched and is transmitted to the offspring, it remains stable
and is propagated in all further cell divisions. This requires that
the previous parental imprinting mark can be erased and that the imprinting
mark, when present, leads to silencing of the transcription of
the imprinted gene. The molecular mechanism that best fits all these
requirements is methylation of cytosine residues at the promoter region
of genes (Fig. 8), and it has indeed been shown that genes in imprinted loci in the mouse
and human genome have reversible methylation of promoter regions on
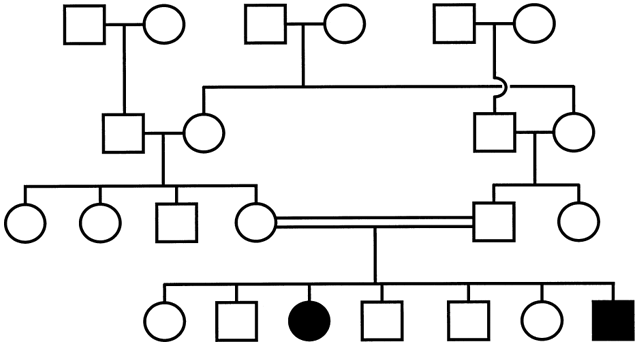
one of their two alleles and that the methylated allele is silenced.8,9,10,11 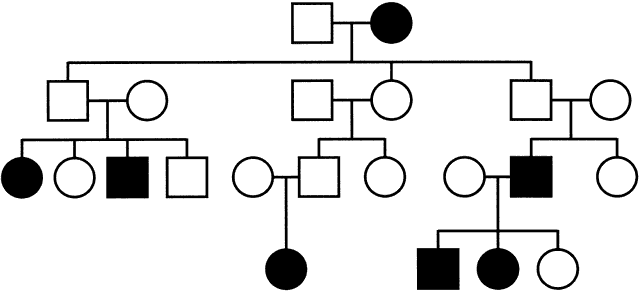 Fig. 7. Pedigree of a maternally imprinted disorder. Note the absence of a phenotype
with transmission of the mutation through the maternal germline
resulting in the presence of unaffected male and female carriers in each
generation. Fig. 7. Pedigree of a maternally imprinted disorder. Note the absence of a phenotype
with transmission of the mutation through the maternal germline
resulting in the presence of unaffected male and female carriers in each
generation.
|
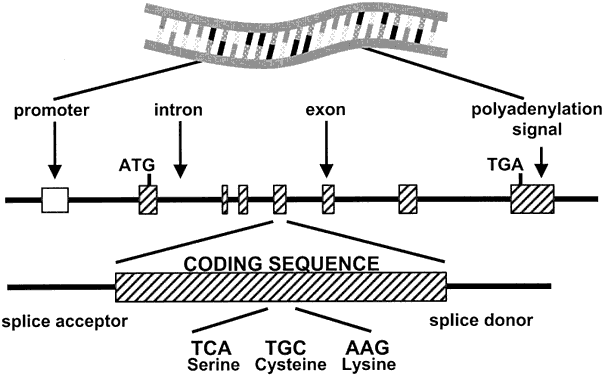
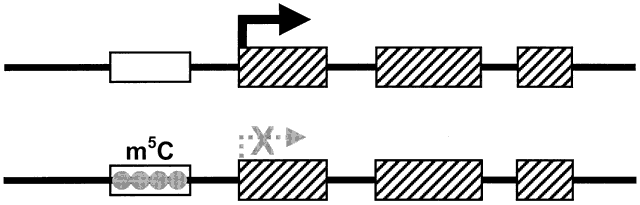 Fig. 8. Methylation of cytosine at promoters of genes leads to transcriptional
silencing of imprinted alleles. The hatched boxes represent exons. White
boxes represent the promoter regions; gray circles represent the methylation
of cytosine residues (m5 C) of one allele, which results in silencing of the downstream gene ( hatched arrow with X ). The thick arrow indicates active transcription of the other allele. Fig. 8. Methylation of cytosine at promoters of genes leads to transcriptional
silencing of imprinted alleles. The hatched boxes represent exons. White
boxes represent the promoter regions; gray circles represent the methylation
of cytosine residues (m5 C) of one allele, which results in silencing of the downstream gene ( hatched arrow with X ). The thick arrow indicates active transcription of the other allele.
|
The number of genes discovered to be imprinted is growing.11 Most, but certainly not all, are grouped together in specific regions
of the genome (imprinting clusters) that usually contain both maternally
and paternally imprinted genes interspersed with genes that are biallelically
expressed. This strongly suggests that a common regulatory
mechanism regulates parental-specific expression of these groups of genes.9 The functional role of imprinting in mammals is unclear, but the favored
explanation is Haig's parental tug-of-war hypothesis. This hypothesis
stems from the observation that most paternally expressed genes
are growth-promoting, whereas maternally expressed genes restrict growth. Among
polygamous species, females must preserve their resources for
multiple litters from different partners and so benefit from smaller
progeny. Males, conversely, benefit from having larger (and thus more
resilient) offspring.8,9,10,11 UNIPARENTAL DISOMY. Imprinting of genes is responsible for disease phenotypes seen in uniparental
disomy (UPD). This is the inheritance in the offspring of both
homologues of a chromosome pair from the same parent (maternal UPD when
both homologues are inherited from the mother, and paternal UPD when
both homologues are inherited from the father). Different genetic mechanisms
can lead to UPD. Chromosome nondisjunction leading to a trisomic
conceptus is sometimes followed by loss of a chromosome or “trisomy
rescue” and is the most commonly observed mechanism. When
the rescue is incomplete, confined placental mosaicism (i.e., the presence of both disomic and trisomic cell lines in the same placenta) can
result; this is occasionally associated with fetal mosaicism
and, in theory, carries a 30% chance of UPD for the involved chromosome
if the chromosome from the parent that only contributes one of the
three is lost (Fig. 9). Alternative mechanisms, such as chromosome duplication in monosomy and
gamete complementation (diploid gamete from one parent is fertilized
by a gamete lacking the same chromosome), can occur.1,2,12 UPD exerts no noticeable consequences when it involves chromosomes without
imprinting effects. If the chromosomes contain imprinted genes, however, UPD
can result in overexpression or underexpression of these genes. For
example, up to 30% of cases of AS result from paternal UPD. 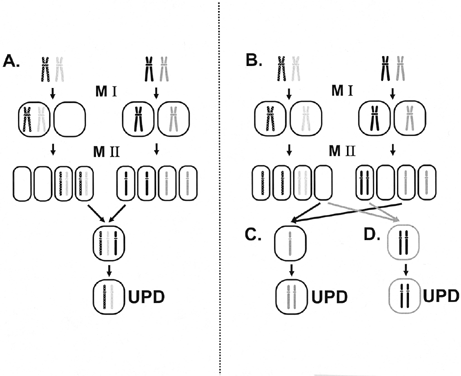 Fig. 9. Mechanisms underlying uniparental disomy. A. Uniparental heterodisomy. Nondisjunction of a chromosome in meiosis I
leads to heterodisomic gametes (two different homologues). A trisomic
zygote is present after fertilization, and trisomy rescue results in the
loss of one homologue. If the homologue from the parent contributing
only one chromosome is lost, uniparental heterodisomy results. B. Uniparental isodisomy usually results from meiosis II nondisjunction, which
results in isodisomic gametes (two identical homologues) as well
as nullisomic gametes (absence of the involved homologue). If a normal
gamete fertilizes a nullisomic gamete, chromosome duplication can occur ( C ). Note that this mechanism can also operate after meiosis I nondisjunction. Fertilization
of a nullisomic gamete by a disomic gamete also results
in uniparental isodisomy ( D ). Fig. 9. Mechanisms underlying uniparental disomy. A. Uniparental heterodisomy. Nondisjunction of a chromosome in meiosis I
leads to heterodisomic gametes (two different homologues). A trisomic
zygote is present after fertilization, and trisomy rescue results in the
loss of one homologue. If the homologue from the parent contributing
only one chromosome is lost, uniparental heterodisomy results. B. Uniparental isodisomy usually results from meiosis II nondisjunction, which
results in isodisomic gametes (two identical homologues) as well
as nullisomic gametes (absence of the involved homologue). If a normal
gamete fertilizes a nullisomic gamete, chromosome duplication can occur ( C ). Note that this mechanism can also operate after meiosis I nondisjunction. Fertilization
of a nullisomic gamete by a disomic gamete also results
in uniparental isodisomy ( D ).
|
The best-known examples of imprinting disorders are PWS and AS and exemplify
well the contribution of the various mechanisms that can bring out
phenotypes associated with imprinting.1,2,8 These two very different developmental disorders result in most instances
from identical interstitial cytogenetic deletions on chromosome
15q11–13. In AS, it is the maternal chromosome that has the deletion, whereas
in PWS, it is the paternal chromosome. AS is characterized
by severe mental retardation, seizures, absence of speech, ataxic gait, hand
flapping, inappropriate laughter, and protruding tongue. Seventy
percent of patients with AS have a maternal deletion, whereas only 3% to 5% have
UPD15 with maternal deficiency, and 4% to 6% of patients
have point mutations in the UBE3A gene encoding E6-AP ubiquitin protein ligase. This gene is biallelically
expressed in most tissues but is paternally imprinted in specific brain
regions and illustrates that genetic imprinting can be tissue-specific.13 PWS children have mild-to-moderate mental retardation, hypotonia with
growth delay followed by hyperphagia and obesity, hypogonadism, and short
stature. It is now known that 65% of PWS results from paternal deletions, whereas
almost all the remaining cases of PWS are due to maternal
UPD. IMPRINTING IN DEVELOPMENT AND HUMAN REPRODUCTION. Triploid conceptuses and the genomes of hydatidiform moles and ovarian
teratomas illustrate well the differences between maternal and paternal
genomes. The majority of complete hydatidiform moles have a normal
karyotype (46,XX), but the whole genome is of paternal origin. This leads
to excessive and abnormal placental development and lack of fetal
development. Ovarian teratomas are thought to result from the parthenogenetic
activation of unfertilized oocytes, and their genome is thus entirely
maternally derived. They can contain a variety of more-or-less
differentiated tissues but never contain trophoblast. When the extra
haploid set of chromosomes of a triploid conceptus is paternally derived, it
results in partial hydatidiform mole and reduced fetal development. The
converse case, when the extra haploid set is maternal in origin, results
in small, underdeveloped placentas.10 Mitochondrial Inheritance Cells contain, on average, 103 to 104 mitochondria, each having two to 10 copies of a 16,569 base-pair long
double-stranded circular DNA molecule. The mitochondrial genome has 37 genes
that encode for a number of proteins of the oxidative phosphorylation
chain, for 22 tRNA molecules and rRNAs. All other proteins are
encoded in the nuclear genome and imported into the mitochondria. The
replication and transcription of mitochondria depend on nuclear-encoded
enzymes and transcription factors.14,15 Virtually all (99.99%) mitochondrial DNA is inherited from the mother. The
oocyte has a large number (106) of mitochondria, whereas most of the already-depleted supply of mitochondria
in the fertilizing sperm is eliminated during the first cell divisions.16 It has been shown that even after intracytoplasmic sperm injection, few
if any paternal mitochondria are retained in the developing embryo. Mitochondrial
disorders follow two patterns of inheritance: mutations
in nuclear-encoded genes follow traditional patterns (AD, AR, XL), whereas
mutations in mitochondrial genes follow typical maternal inheritance. Mitochondria
divide and replicate their DNA asynchronously from the
meiotic and mitotic cell divisions, resulting in a variable number
in each cell. In general, all mitochondria of an individual have the same
genome (homoplasmy). Mutations in mitochondrial genes can affect all (homoplasmic
mutations) or a subset of the mitochondria in each cell (heteroplasmic
mutations); the latter leads to variations in the phenotype
between individuals within the same family. During oogenesis and/or
in the earliest cell divisions after fertilization, a subset of the
mitochondria present in the oocyte is selectively amplified, leading
to a “mitochondrial bottleneck” (Fig. 10). The fraction of mutated mitochondria in this amplified population results
in different levels of heteroplasmy in the offspring. The pedigree
of a typical mitochondrial disorder is given in Figure 11 and illustrates the absence of paternal transmission, with equal maternal
transmission to affected sons and daughters.14,15 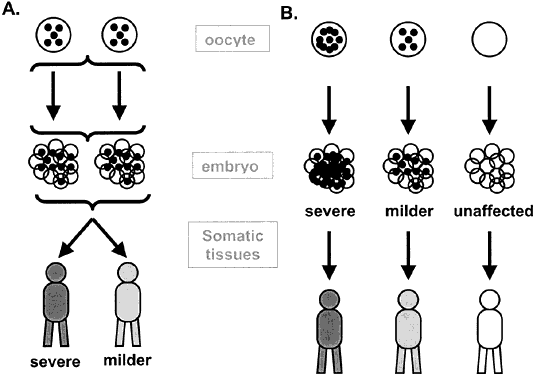 Fig. 10. The mitochondrial bottleneck. Selective sampling of a subset of mutated
mitochondria ( black circles) during early embryonic development leads to variable ratios of wild-type
and mutated mitochondria in the offspring and variable degrees of
phenotypic severity. It is unclear whether this sampling occurs primarily
in later embryonic cell divisions ( A) or in the earliest divisions of oogenesis and embryo development ( B ). Fig. 10. The mitochondrial bottleneck. Selective sampling of a subset of mutated
mitochondria ( black circles) during early embryonic development leads to variable ratios of wild-type
and mutated mitochondria in the offspring and variable degrees of
phenotypic severity. It is unclear whether this sampling occurs primarily
in later embryonic cell divisions ( A) or in the earliest divisions of oogenesis and embryo development ( B ).
|
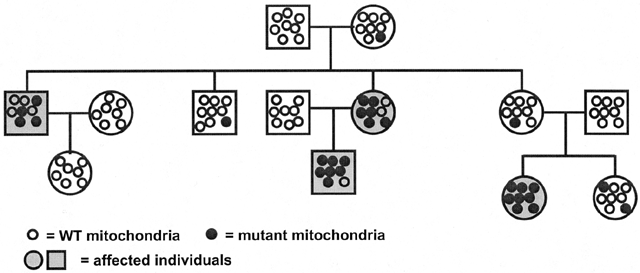 Fig. 11. Pedigree with mitochondrial inheritance and heteroplasmy. Note the absence
of paternal transmission and equal maternal transmission to males
and females. Heteroplasmy leads to the presence of unaffected transmitting
females. Fig. 11. Pedigree with mitochondrial inheritance and heteroplasmy. Note the absence
of paternal transmission and equal maternal transmission to males
and females. Heteroplasmy leads to the presence of unaffected transmitting
females.
|
Mitochondrial disorders are subdivided into three classes. Class I disorders (e.g., Leigh syndrome) are caused by mutations in nuclear genes and have traditional
Mendelian patterns of inheritance. Class II disorders result
from point mutations in mitochondrial genes; are usually maternally inherited; and
often cause muscular, neurologic, and eye phenotypes (e.g., myoclonic epilepsy with lactic acidosis and stroke; neurogenic weakness, ataxia, retinitis
pigmentosa, myoclonic epilepsy with ragged red fibers). Class
III mutations are mostly sporadic-occurring mitochondrial
deletions and duplications that can cause a spectrum of diseases (e.g., diabetes and deafness, Pearson syndrome, Kearns-Sayre syndrome). The
accumulation of free oxygen radicals in mitochondria recently has been
proposed to play a role in aging and possibly in neurodegenerative disorders
such as Parkinson's disease, Alzheimer's disease, and
Friedreich's ataxia.14,15 MOLECULAR DIAGNOSIS AND PRENATAL DETECTION OF MITOCHONDRIAL DISORDERS. Specific point mutations for many class II mitochondrial diseases are
known and detectable, but the sensitivity of the test depends on the degree
of heteroplasmy and the specific tissue examined. This makes prenatal
genetic counseling and molecular testing in these conditions extremely
challenging, as the mitochondrial content of amniocytes or chorion
villi is often not representative of the organs that are commonly
affected (e.g., brain, muscle, heart). Interested patients should be informed about these
shortcomings and referred to specialized genetic counselors.17,18 |