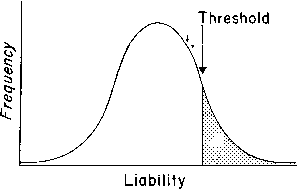
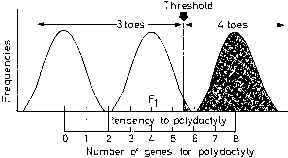
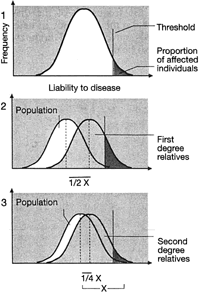
Familial resemblances for anatomic characteristics are influenced by not one but several genes that cumulatively can produce enough genotypes to mimic a normal distribution in the general population. To illustrate this, let us consider the result of sequentially increasing the number of genes influencing a given trait.
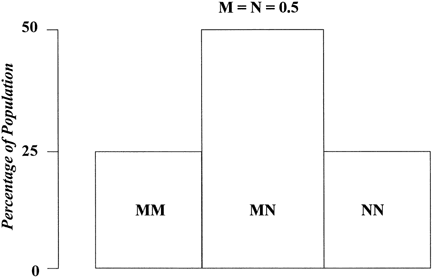
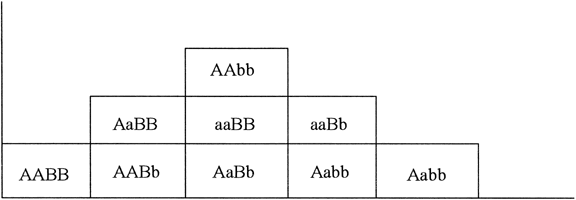
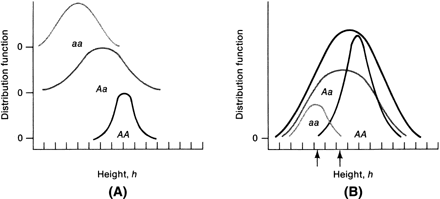
Suppose only one gene controls a given trait and that this gene has only two alleles (A,a). If the frequency of allele A equals the frequency of allele a, 25% of the population is AA based on Hardy-Weinberg equilibrium: p = q = 0.5; p2 = q2 = 0.25; the frequency of aa is also 25%. Aa accounts for 50% (2 pq = 0.50) (Fig. 1). Now suppose that not one but two genes influence a given trait. At the second locus, alleles B and b exist. Nine genotypes are now possible: AABB, AABb, AAbb, AaBB, AaBb, Aabb, aaBB, aaBb, and aabb (Table 1). The population will contain nine phenotypic classes if alleles A, B, a, and b each exert dissimilar influences. If alleles A and B or a and b exert equal effect, only five phenotypic classes exist (Fig. 2). As the number of genes controlling a trait increases, the number of genotypes in the population increases geometrically. If three genes exist, each with two alleles, there are 27 genotypic classes (3n). Even more genotypes exist per locus if the locus has more than two alleles. If there is one locus with three alleles per locus, there are 6 genotypes (Table 2). If there are 2 genes, each with 3 alleles, there are 36 genotypes (see Table 2).
|
No. of Loci | Alleles | Genotypes | No. of Genotypes (Formula) |
1 | (M,N) | MM MN NN | 3(31) |
2 | (M,N;R,S) | MMRR MMRS | 9 (32) |
|
| MMSS MNRR |
|
|
| MNRS MNSS |
|
|
| NNRR NNRS |
|
|
| NNSS |
|
N | (2 alleles per locus) |
| 3n |
Genotypes listed assume two alleles per locus, each of which exerts a differential phenotypic effect.
|
No. of |
|
| No. of |
Loci | Alleles | Genotypes | Genotypes (Formula) |
1 | (M,N,O) | MM MN MO | 6(61) |
|
| NN NO OO |
|
2 | (M,N,O;P,Q,R) |
| 36(62) |
N | (3 alleles per locus) |
| 6n |
Genotypes listed assume three alleles per locus.
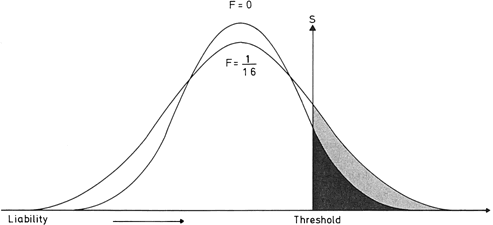
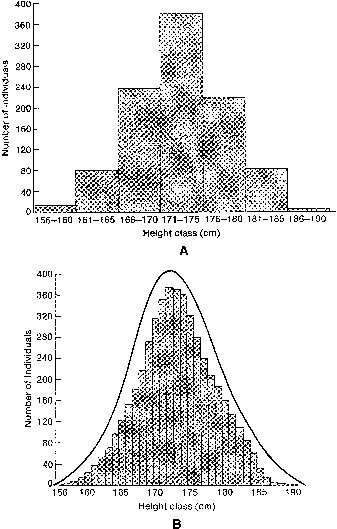
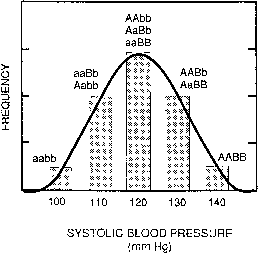
If one histographically represents the number of individuals in each genotypic class, a normal distribution is increasingly approximated as more genotypes exist. With only a few genes, 27 or 36 different genotypes can be produced (see Tables 1 and 2), enough to mimic continuous variation. Figure 3 shows this concept in a different way. If one stratifies heights in the general population into increasingly smaller intervals (1 cm vs 5 cm), histographic representation of the phenotypes better fits the normal distribution. Figure 4 illustrates this for these pathologic traits, the assumption being that only two genes, each with two alleles, determine blood pressure. Table 3 lists several physiologic or anatomic variables for which polygenic inheritance with continuous variation can plausibly be assumed. See Simpson and Elias1 for additional examples.
|
|
Age of menarche
Age of natural menopause (oocyte pool)
Blood pressure during pregnancy
Gestational length
Height
Pelvic shape
Resistance against pelvic relaxation
Responsiveness to medications
Weight (tendency toward obesity)
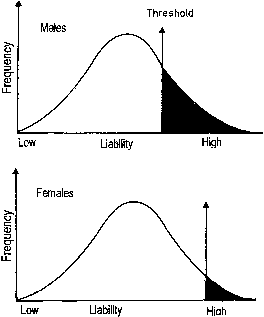
Ostensibly normal distribution of phenotypes in the population can also be explained by several alleles or several genes having nonoverlapping distributions. If only the phenotype is measured, not individual alleles or genes, a normal distribution is seen in the general population (Fig. 5).
|
|
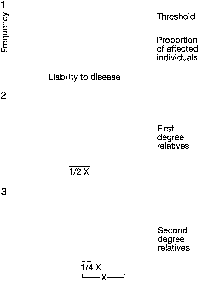
Applying the principles of polygenic inheritance explains why offspring usually, but do not always, reflect parental phenotype. Height can serve as a hypothetical example, for it is well established that a child's height correlates with his/her midparental height, corrected for sex. Suppose height is governed by three genes, each with only two alleles (A,a; B,b;C,c). Each upper case allele (A, B, C) might confer an additional 3 inches in height above some threshold, hypothetically here 60 inches for males and 54 inches for female. Each lower case allele (a, b, c) might contribute nothing above the threshold. Thus, a male of genotype AaBBcC would be 72 inches tall [60 + (4 × 3) = 72]; a female of genotype AaBbCc would be 63 inches tall [54 + (3 × 3) = 63]. On average, one would expect the male parent in the above example to contribute 2 upper case alleles (4/2 = 2) and the female parent 1.5 (3/2 = 1.5). Thus, a child will on average inherit 3.5 upper case alleles and thus approximate parental heights. Offspring, however, could inherit between 1 and 6 upper case alleles and, hence, show heights ranging from 63 to 78 inches in males and 57 to 72 inches in females. The likelihood of various genotypic possibilities producing these extremes is illustrated in Table 4.
|
|
| Parental Heights (inches) | ||
(1) Parental genotypes and height: |
| Male | Aa BB Cc | 72 | |
|
| Female | Aa Bb Cc | 63 | |
|
|
| Offspring (inches) Height | ||
| Genotype in | Probability of | Male | Female | |
| Offspring | Genotype Arising |
|
| |
(2) Probabilities of selected | Aa Bb Cc | 1/2 × 1/2 × 1/2 = 1/8 | 69 | 63 | |
genotypes in offspring and | AA BB CC | 1/4 × 1/2 × 1/4 = 1/32 | 78 | 72 | |
expected height | aa Bb cc | 1/4 × 1/2 × 1/4 = 1/32 | 63 | 57 | |
Baseline height threshold (inches): Males: 60; Females: 54.
Calculation of height above threshold: For each A, B, or C allele add 3;for each a, b, or c allele add 0.
Murphy and Chase,2 Griffiths and associates,3 Vogel and Motulsky,4 and Lynch and Walsh5 provide more detailed and mathematically substantiated discussion of the underlying basis of polygenic inheritance.