Sickle Cell Anemia Sickle cell anemia is an autosomal recessive disorder affecting one out
of 400 African-American children in the United States. The defect results
from a single nucleotide substitution (A to T) in the sixth codon (GAG
to GTG) of the β-globin gene. This mutation leads to the amino
acid substitution of valine for glutamine at position 6 of the β-globin
polypeptide. Previously, diagnosis of sickle cell anemia was
based on the detection of the abnormal type of hemoglobin in fetal blood. However, fetal
blood sampling has become almost obsolete, as DNA-based
diagnosis has developed. The mutation causing sickle cell anemia coincidentally resides within the
recognition sites of restriction enzymes Mst II, CvnI, and Bsu II. Individuals with the sickle cell mutation lack the restriction site
present in individuals with normal β-globin genes. One procedure
for diagnosing fetuses affected with sickle cell anemia using RFLP analysis
is illustrated in Figure 4. Recently, more rapid diagnosis of sickle cell has been possible using
PCR.6 With this method, a 725 bp region that includes the sickle cell mutation
is amplified. The amplified DNA is digested with Bsu II, and then the digested product is separated by electrophoresis. The
gel is stained with ethidium bromide, and the banding patterns can be
directly visualized under ultraviolet light (Fig. 5). This procedure is much faster and more efficient because it eliminates
the need to perform Southern blotting, hybridization with labeled probe, and
autoradiography. This straightforward, direct method of diagnosis
can be applied to any disease in which the mutation causing the
disease removes or creates a restriction site. 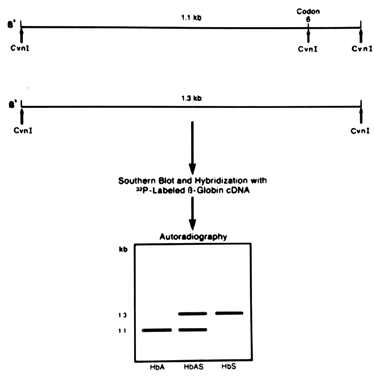 Fig. 4. Use of a radioactively labeled β-globin probe to diagnose sickle cell
anemia. The mutation causing the disease coincides with a Cvn I site. Chromosomes with the sickle mutation lack the site that chromosomes
with normal β-globin genes have. After digestion with Cvn I and hybridization to a β-globin probe, DNA from individuals with
sickle-cell anemia yields a 1.3 kb fragment, DNA from individuals with
two normal β-globin genes yields a 1.1 kb fragment, and carriers
have 1.1- and 1.3-kb fragments. Fig. 4. Use of a radioactively labeled β-globin probe to diagnose sickle cell
anemia. The mutation causing the disease coincides with a Cvn I site. Chromosomes with the sickle mutation lack the site that chromosomes
with normal β-globin genes have. After digestion with Cvn I and hybridization to a β-globin probe, DNA from individuals with
sickle-cell anemia yields a 1.3 kb fragment, DNA from individuals with
two normal β-globin genes yields a 1.1 kb fragment, and carriers
have 1.1- and 1.3-kb fragments.
|
 Fig. 5. Diagnosis of sickle cell anemia in PCR-amplified DNA. Amplified DNA containing
codon 6 of the β-globin gene is digested with Cvn and electrophoresed.6 DNA is labeled with ethidium bromide and visualized
under ultraviolet light. The sickle mutation eliminates the recognition
sequence of this enzyme. Therefore, the HbS allele is visualized
as a 340-bp band and the HbA allele as 200- and 140-bp bands. A constant
band of 100 base pair is present in all individuals. Fig. 5. Diagnosis of sickle cell anemia in PCR-amplified DNA. Amplified DNA containing
codon 6 of the β-globin gene is digested with Cvn and electrophoresed.6 DNA is labeled with ethidium bromide and visualized
under ultraviolet light. The sickle mutation eliminates the recognition
sequence of this enzyme. Therefore, the HbS allele is visualized
as a 340-bp band and the HbA allele as 200- and 140-bp bands. A constant
band of 100 base pair is present in all individuals.
|
Cystic Fibrosis Cystic fibrosis (CF) is the most common autosomal recessive genetic disorder
in the northern European population. Approximately one in 25 Caucasians
are carriers (i.e., heterozygotes) for the gene, and one child in every 2500 births is affected
with CF. Affected individuals rarely live past their thirties
and suffer from debilitating pulmonary and digestive disorders throughout
their lives. By demonstrating linkage between CF and RFLPs on chromosome 7, the
CF gene was mapped to this chromosome in 1985,7, 8, 9, 10 but only recently was the CF gene itself identified and sequenced.11, 12, 13 A three-base pair deletion, resulting in the loss of a phenylalanine residue
at position 508 (called delta F508), was found in 75% of CF chromosomes
studied by Kerem and associates. The remainder of CF chromosomes
each carry one of many (more than 100) less common mutations. The direct detection of the delta F508 mutation does not require studying
a living affected relative with CF and could be used to screen the
general population for CF carrier status.13 Because only approximately 75% of CF carriers have the delta F508 mutation, however, 25% of
carriers would go undetected if we screened for
this mutation only. As a result, 25% of true carriers (or approximately 1% of
individuals screened) will have a negative test, but will in truth
be CF carriers. To further complicate matters, the frequency of the
delta F508 mutation is lower in ethnic and racial groups other than
northern European, non-Ashkenazi Caucasian populations. In these groups (such
as African Americans, Ashkenazi Jews, southern and eastern Europeans), the
frequency varies from 30% to 60%. Due to these limitations, screening
for CF mutations is recommended currently only for relatives
of individuals with CF and for spouses of known CF carriers. Screening
for CF-carrier status in the general population is not recommended
until additional mutations can be tested for, thereby reducing the
false-positive rate and increasing the sensitivity of the screen.14, 15 It is anticipated that these problems will be overcome in the near future, and
screening for CF carriers will become available at genetic centers. Direct testing for delta F508 or other known mutations is useful in many
families with a CF child. Because not all CF carriers have mutations
that can be detected, however, linkage analysis using RFLPs is still
required in some families. One method for detecting the delta F508 mutation
utilizes PCR, dot blotting the amplified product, and hybridization
to sequence-specific oligonucleotide (SSO) probes (Fig. 6).16 This method quickly determines whether an individual carries zero, one, or
two copies of this mutation. 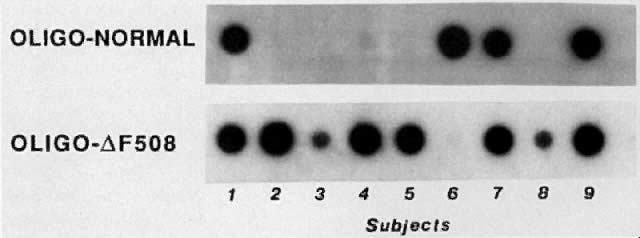 Fig. 6. Direct detection of the delta F508 mutation in DNA from nine children with
CF. DNA is amplified using PCR and placed onto a membrane in duplicate
as “dot blots.” Each blot is then hybridized to one of
two SSOs. The nucleotides in the first SSO (oligo-N) are complementary
to the DNA in the nondeleted gene ( i. e ., the normal sequence); the sequence of the second SSO (oligodelta F508) is
complementary to the sequence with the deletion. DNA from individuals
homozygous for the deletion hybridizes only to the second SSO, DNA
from individuals heterozygous for the deletion hybridizes to both
oligos, and DNA from individuals homozygous for the normal sequence hybridizes
only to the first SSO. In the figure, five children are homozygous
for the mutation, and three are heterozygous for the mutation. These
three children presumably have a nondelta F508 mutation on their
other chromosome. One child lacks the delta F508 mutation and presumably
has nondelta F508 mutations on both chromosomes. Fig. 6. Direct detection of the delta F508 mutation in DNA from nine children with
CF. DNA is amplified using PCR and placed onto a membrane in duplicate
as “dot blots.” Each blot is then hybridized to one of
two SSOs. The nucleotides in the first SSO (oligo-N) are complementary
to the DNA in the nondeleted gene ( i. e ., the normal sequence); the sequence of the second SSO (oligodelta F508) is
complementary to the sequence with the deletion. DNA from individuals
homozygous for the deletion hybridizes only to the second SSO, DNA
from individuals heterozygous for the deletion hybridizes to both
oligos, and DNA from individuals homozygous for the normal sequence hybridizes
only to the first SSO. In the figure, five children are homozygous
for the mutation, and three are heterozygous for the mutation. These
three children presumably have a nondelta F508 mutation on their
other chromosome. One child lacks the delta F508 mutation and presumably
has nondelta F508 mutations on both chromosomes.
|
In CF families in which only one or neither parent carries a mutation that
can be detected, prenatal diagnosis relies on family-based linkage
studies. Figure 7A illustrates the relationship between the CF gene and a closely linked RFLP, KM. 19, in a family with three affected and two unaffected children. The pedigree
of the family and RFLP banding patterns after electrophoresis of
PCR-amplified DNA that was digested with the restriction enzyme, PstI, is shown in Figure 7B. The parents (I.1 and I.2) are presumed to be heterozygous carriers of
the CF gene because they have three affected children. The affected children
are assumed to be homozygous for the CF gene. The unaffected children
can be either heterozygous carriers of the CF gene or can inherit
normal genes from both parents and be homozygous normal. After DNA
analysis, it is determined that both parents are heterozygous for the KM. 19 polymorphism (i.e., heterozygous for the presence of the restriction site; genotype +,-). The
three affected children (II.1, II.3, II.5) are homozygous for
the presence of the restriction site (genotype, +,+). Thus, we
can deduce that each affected child inherited the chromosome containing
the + allele from both parents, and the CF gene must be on
these parental chromosomes. Both parental chromosomes with the - allele
must therefore carry the normal gene. We can further deduce that the
brother with genotype, -,- (II.2), inherited the normal gene from each
parent and is not a carrier for CF. The sister with genotype +,- (II.4), inherited
the chromosome with the normal gene from one parent
and the chromosome with the CF gene from the other parent and is presumed
to be a CF carrier.17 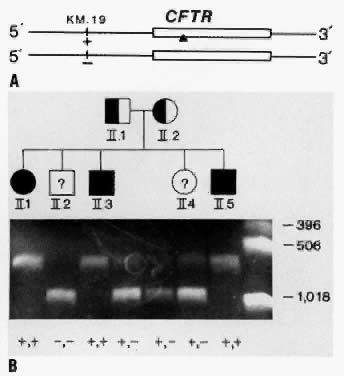 Fig. 7. A. The relationship between the CFTR gene and a closely linked RFLP, KM.19, on chromosome 7. In this example, one chromosome has the + allele
at the KM.19 locus (presence of a restriction site), and one chromosome has the - allele
at this locus (absence of a restriction site). The + allele
at the KM.19 locus is on the same chromosome as the abnormal CFTR gene (designated
by a solid triangle) and the - allele is on the same chromosome as the
normal CFTR gene. The KM.19 alleles can be used to track the inheritance of the CFTR in families. B. Diagnosis of cystic fibrosis using the KM.19 RFLP in PCR-amplified DNA in a family with three affected children. After
PCR, DNA was digested with restriction enzyme Pst I and electrophoresed. DNA is labeled with ethidium bromide and visualized
under ultraviolet light. Chromosomes that lack the Pst I cutting site (-) appear as a single 950 bp band. Chromosomes that have
the cutting site (+) appear as 650 and 300 bp bands. The smallest 300 bp
band cannot always be seen in the heterozygote. Fig. 7. A. The relationship between the CFTR gene and a closely linked RFLP, KM.19, on chromosome 7. In this example, one chromosome has the + allele
at the KM.19 locus (presence of a restriction site), and one chromosome has the - allele
at this locus (absence of a restriction site). The + allele
at the KM.19 locus is on the same chromosome as the abnormal CFTR gene (designated
by a solid triangle) and the - allele is on the same chromosome as the
normal CFTR gene. The KM.19 alleles can be used to track the inheritance of the CFTR in families. B. Diagnosis of cystic fibrosis using the KM.19 RFLP in PCR-amplified DNA in a family with three affected children. After
PCR, DNA was digested with restriction enzyme Pst I and electrophoresed. DNA is labeled with ethidium bromide and visualized
under ultraviolet light. Chromosomes that lack the Pst I cutting site (-) appear as a single 950 bp band. Chromosomes that have
the cutting site (+) appear as 650 and 300 bp bands. The smallest 300 bp
band cannot always be seen in the heterozygote.
|
Prenatal diagnosis of CF in subsequent pregnancies in this family is also
possible. Fetal DNA derived from chorionic villi or from amniotic fluid
cells can be analyzed using the methods described above. As previously
discussed, the major potential source of error in linkage studies
is the probability of recombination between the restriction site and
the abnormal gene in the parents' gametes. Thus, results from indirect
DNA testing are given as a probability. For example, KM. 19 is within 100 kbp of the CF gene. Thus, the recombination frequency between
the KM. 19 gene and the CF gene, determined from family studies, is small (less than 1%). If
prenatal testing of a subsequent pregnancy in the couple in Figure 7 revealed the +,- genotype, the couple should be counseled that the
probability that the fetus will have CF is equal to the probability that
recombination occurred between the CF gene and the RFLP in the chromosome
with the - allele (<1%). On the other hand, if the fetus was
genotype, +,+, the probability that it will have CF is >99%. If
the probability based on recombination calculations proves too
uncertain, or for families in whom DNA diagnosis is not informative, measurement
of amniotic fluid microvillar intestinal enzymes (i.e., alkaline phosphatase, γ-glutamyl transpeptidase, leucine amniopeptidase) may
be useful, albeit not 100% sensitive or specific.18 Congenital Adrenal Hyperplasia (CAH) Congenital adrenal hyperplasia (CAH) is an autosomal recessive disorder
of cortisol biosynthesis, which is caused in 95% of cases by a deficiency
in the enzyme steroid 21-hydroxylase.19 This enzyme is required for the conversion of progesterone and 17-hydroxyprogesterone
to 11-deoxycorticosterone and 11-deoxycorticosterol, which
are intermediate products in mineralocorticoid and glucocorticoid
biosynthesis, respectively. Due to a lack of feedback in CAH suppression
of this pathway, there is a compensatory increase in ACTH, leading
to adrenal hyperplasia and excessive secretion of precursor steroids. The
increased secretion of adrenal androgens (such as DHEA and DHEAS) leads
to increased conversion of these products to testosterone and dihydrotestosterone. CAH occurs in a number of forms, ranging from mild to severe. The milder
forms often present during childhood. More attenuated forms present
at puberty (or even after puberty), with menstrual irregularities and
infertility. The severe form is characterized by early virilization in utero, leading to marked masculinization of the external genitalia. Affected
females may, in fact, be mistaken for males at birth, with bilateral
cryptorchidism and hypospadias. The diagnosis may not be as obvious in
males because the external genitalia are normal. Unrecognized salt wasting
in neonates with CAH is often fatal because the inadequacy of glucocorticoids
can lead to vascular collapse, shock, and death. The severe
form of CAH occurs with a frequency of one in five to 10,000 births, and
the milder (or attenuated) forms occur at a frequency of approximately
one in 1000 births.20 The virilization process in utero begins early in pregnancy because the genital ridge forms at 9 to 10 weeks
of gestation. Fortunately, the virilization process of female fetuses in utero can be inhibited by treatment of the mother during pregnancy with dexamethasone, thereby
eliminating the need for extensive corrective surgery
after birth. Because steroid treatment of the mother is not totally
benign, however, treatment should be discontinued if the fetus is determined
to be either homozygous or heterozygous for the normal 21-hydroxylase
genes, or if the fetus is male. Thus, early and correct diagnosis
of this genetic disease is of great importance. However, prenatal
detection of CAH using biochemical tests for increased concentrations
of amniotic fluid 17-hydroxyprogesterone are often inconclusive and are
usually only feasible after 13 to 14 weeks' gestation. The gene encoding the enzyme 21-hydroxylase has been mapped to the short
arm of chromosome 6, positioned between the genes encoding HLA-B and
HLA-DR.21, 22, 23 Gene probes for the 21-hydroxylase gene have been developed and molecular
genetic studies of this region have demonstrated that two copies of
the 21-hydroxylase gene (called A and B) are present in this region.23, 24 These two genes can be differentiated by hybridizing 21-hydroxylase gene
probes to TaqI digested DNA: The A gene is detected on a 3.2 kb fragment, whereas the
B gene resides on a 3.7 kb fragment. Detailed sequence analysis of the
two genes has revealed that they are >90% homologous. Three deleterious
mutations in the A gene render it nonfunctional, whereas the B
gene is a functional gene, encoding the enzyme.25, 26 Analysis of southern blots of genomic DNA from patients with salt wasting
CAH has revealed that, in about 25% of patients, the 21-hydroxylase
B gene is either deleted or converted to the A gene.27 In these families, a direct diagnosis can be made by determining the presence
or absence of the 3.7 kb fragment. In most cases, however, patients
with CAH have the 3.7 kb TaqI fragment (B gene). In these families, the prenatal diagnosis of CAH depends
on the indirect method, even though the biochemical defects for
this disorder are known. RFLPs detected by probes for the HLA-B or HLA-DRβ loci
have proven particularly useful for linkage studies in
CAH families.
The author was supported in part by NIH research grant HD21244. |
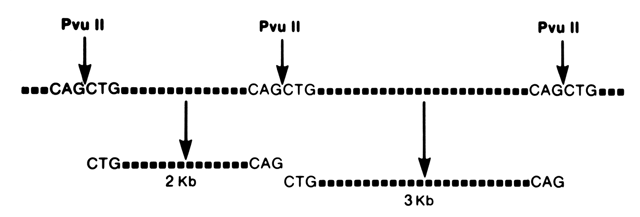

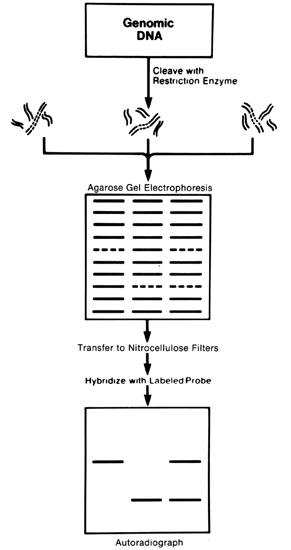
 25 base pairs) that is synthesized in the laboratory. The usefulness of
these short sequences is that, under particular conditions, the probe
will not hybridize unless the nucleotides in the DNA being tested exactly matches the nucleotide sequence in the probe. Thus, an oligonucleotide
probe will differentiate between sequences that differ by a single base
pair (e.g., sickle vs normal β-globin gene). A major limitation of PCR is that
the DNA sequence on both sides of the mutation (i.e., the flanking sequences) must be known. However, as this limitation is
overcome with more information about disease genes, analyses utilizing
PCR are becoming the method of choice.
25 base pairs) that is synthesized in the laboratory. The usefulness of
these short sequences is that, under particular conditions, the probe
will not hybridize unless the nucleotides in the DNA being tested exactly matches the nucleotide sequence in the probe. Thus, an oligonucleotide
probe will differentiate between sequences that differ by a single base
pair (e.g., sickle vs normal β-globin gene). A major limitation of PCR is that
the DNA sequence on both sides of the mutation (i.e., the flanking sequences) must be known. However, as this limitation is
overcome with more information about disease genes, analyses utilizing
PCR are becoming the method of choice.