Formerly known as “testicular feminization syndrome,” androgen
insensitivity syndrome (AIS) is an X-linked disorder in which a 46,XY
shows a female phenotype. The prevalence of complete AIS has been
reported to be 1 in 60,000.10,13 Diagnosis is usually not made until puberty, at which time normal linear
growth and normal breast development have occurred, but menarche has
not. Despite pubertal feminization, some persons with androgen insensitivity
show clitoral enlargement and labioscrotal fusion. The term partial (incomplete) androgen insensitivity (formerly incomplete testicular
feminization) is applied to these patients. Both complete and partial
androgen insensitivity are inherited in an X-linked recessive fashion, and
both involve the same gene; however, the two disorders are considered
distinct because they clearly breed true in a given family. Complete Androgen Insensitivity Syndrome Persons with complete AIS may be quite attractive and show excellent breast
development, and most are similar in appearance to unaffected females
in the general population. Their breasts contain normal ductal and
glandular tissue, but their areolae are often pale and underdeveloped. Pubic
and axillary hair are usually sparse, but scalp hair is normal. The
vagina terminates blindly. Sometimes vaginal length is shorter
than usual, presumably because müllerian ducts fail to contribute
to formation of the vagina. Rarely, the vagina is only 1 to 2 cm long
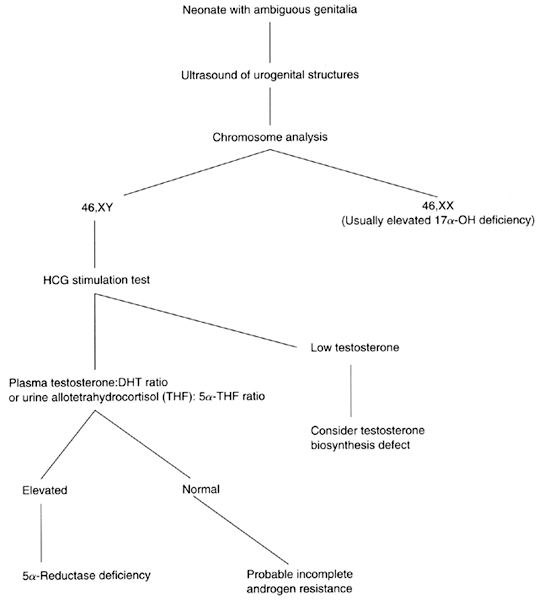
or represented merely by a dimple (Fig. 6).30  Fig. 6. Photograph of a patient with complete androgen insensitivity syndrome.(Courtesy Charles B. Hammond, Duke University. From Simpson JL: Disorders
of Sexual Differentiation: Etiology and Clinical Delineation. New York, Academic
Press, 1976) Fig. 6. Photograph of a patient with complete androgen insensitivity syndrome.(Courtesy Charles B. Hammond, Duke University. From Simpson JL: Disorders
of Sexual Differentiation: Etiology and Clinical Delineation. New York, Academic
Press, 1976)
|
Neither a uterus nor fallopian tubes are ordinarily present. Occasionally, fibromuscular
remnants, rudimentary fallopian tubes, or rarely even
a uterus are detected.31,32,33 The absence of müllerian derivatives is expected because anti müllerian
hormone, which is secreted by the fetal Sertoli cells, is
not an androgen; therefore, müllerian regression is expected to occur
in males with androgen sensitivity, just as in normal males. The
only other condition in which a uterus is absent in a phenotypic female
is müllerian aplasia, which is readily distinguishable on the basis of pubic hair and a 46,XX
complement. In complete AIS, testes are usually normal in size and located in the abdomen, inguinal
canal, or labia (i.e., anywhere along the path of embryonic testicular descent). If present
in the inguinal canal, testes can produce inguinal hernias. It may therefore
be worthwhile to determine cytogenetic status of prepubertal girls
with inguinal hernias, although most will be 46,XX. Height is slightly increased over that of normal women, but unremarkable
compared with 46,XY males. Presumably the increased height reflects
the influence of the Y chromosome. Consistent with this is the impression
expressed by many clinicians that the hands and feet of these women
are relatively large compared with those of normal women. The frequency of gonadal neoplasia is increased, but probably less so than
once believed. In 1953, Morris and Mahesh34 stated that 22% of affected patients had neoplasia. The actual risk is
probably no greater than 5%.35,36 Most investigators now agree that the risk of neoplasia is low before 25 to 30 years
of age. Benign tubular adenomas (Pick's adenomas) are
especially common in postpubertal patients, probably as result of
increased secretion of luteinizing hormone. The pathogenesis of complete
AIS involves end-organ insensitivity to androgens. HORMONE LEVELS. Serum testosterone and dihydrotestosterone levels are normal or elevated
in AIS. Luteinizing hormone and estrogen are elevated, suggesting abnormal
gonadal-hypothalamic feedback. Consistent with this, Leydig cells
are hyperplastic.18 Androgen binding is absent, decreased, or qualitatively abnormal. In a
study of androgen binding of genital skin fibroblasts from 42 patients
with complete AIS, 24 (57%) showed absent androgen binding, 15 (36%) decreased
or qualitatively abnormal androgen binding, and 3 (7%) ostensibly
normal androgen binding.8 GENETICS. AIS is inherited as an X-linked recessive disorder, the result of a defect
in the androgen-receptor gene. Chromosome analysis is 46,XY. 23 The androgen-receptor gene was cloned by several groups in 1988, and is
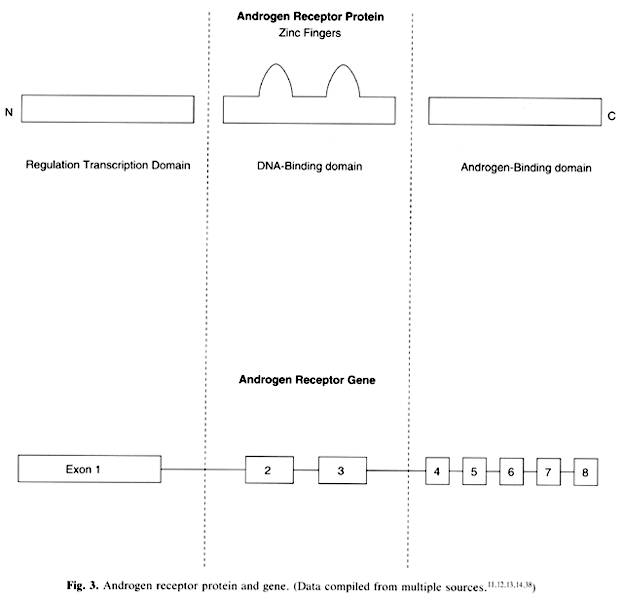
located on the X chromosome at Xq11–12.37 The gene is 90 kb in length and consists of eight exons that encode the
three domains of the receptor (see Fig. 3).11,12,13,14,38 Exon 1 encodes the transcription regulation domain. This region includes
polymorphic CAG repeats, which aid in restriction fragment length polymorphism (RFLP) diagnosis. Exons 2 and 3 encode the DNA-binding domains; exons 4 to 8 encode
the androgen-binding domains. Many mutations
involving the androgen-receptor gene have been identified.39,40,41,42,43 Like most genes, molecular heterogeneity exists among affected persons. Surprisingly, however, both receptor-positive and receptor-negative
persons with complete AIS seem indistinguishable clinically. Approximately 70% of cases studied have shown a mutation in the androgen-receptor
gene. Deletions are rare, as are insertions.44 More commonly encountered are point mutations involving single nucleotide
changes that result in either substitution of an unscheduled amino
acid, deletion of three nucleotides with preservation of an open reading
frame, or generation of an unscheduled stop codon to cause premature
message termination of the message and production of a nonfunctional
protein. Large deletions and mutations that result in premature termination (stop
codon) yield no functional receptor and predictably cause
complete AIS.45 Point mutations resulting from single nucleotide substitutions might be
associated with the production of some androgen receptors. Sometimes
the receptor may be unstable or characterized by poor binding.45 Clarification of the relationship between phenotype and specific androgen-receptor
gene mutations is under way, with an emphasis on an understanding
of the consequences of altered androgen-receptor kinetics on tertiary
structure.46,47,48,49 Especially interesting are circumstances in which substitution resulting
in one new amino acid produces complete AIS,43 but substitution of another produces only partial AIS. An example was
reported by Kazemi-Esfarjani and associates48 in which valine-865 was substituted with either methionine or leucine, causing
complete or partial AIS, respectively. Studies such as this have
become increasingly important both academically and clinically because
of the marked molecular heterogeneity of AIS, which has made it difficult
to improve our diagnostic capability in sporadic cases. Of course
various strategies (e.g., direct mutation detection, RFLP analysis) can still be devised to detect
heterozygotes or hemizygotes once a mutant sequence is identified
in a given family.49 DIAGNOSIS. Patients with AIS have normal female external genitalia, feminize at puberty (develop
breasts), and show primary amenorrhea. History may elicit
prior inguinal hernias. Physical examination may reveal a shortened
and blindly ending vagina, as well as an absent uterus and cervix (Fig. 7).50 An absent uterus and ovaries is found on ultrasound examination, and chromosome
analyses reveal a 46,XY complement. Receptor studies or DNA
studies are not routinely available except through laboratories involved
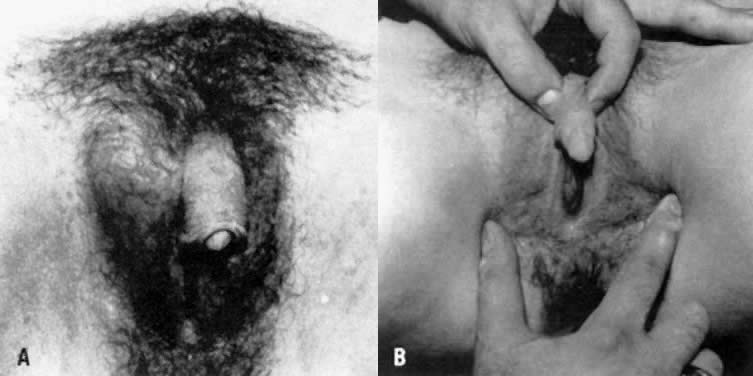
with AIS research, and thus are not considered obligatory. 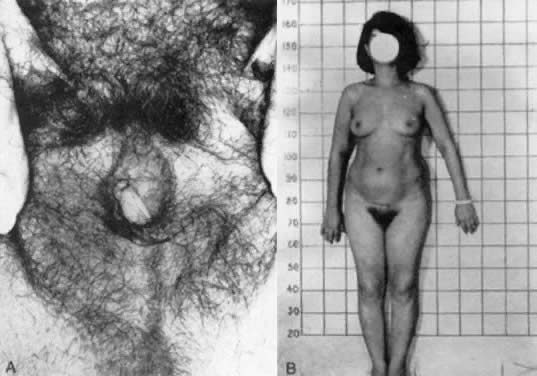 Fig. 7. A and B. Patient with partial androgen insensitivity syndrome.(Park IJ, Jones HW: Familial male hermaphroditism with ambiguous external
genitalia. Am J Obstet Gynecol 108:1197, 1970) Fig. 7. A and B. Patient with partial androgen insensitivity syndrome.(Park IJ, Jones HW: Familial male hermaphroditism with ambiguous external
genitalia. Am J Obstet Gynecol 108:1197, 1970)
|
Prenatal diagnosis for AIS can be achieved by the use of DNA obtained from
chorionic villi sampling or amniocentesis. Direct mutation analysis
or RFLP analysis can be used if the molecular defect is known.44 MANAGEMENT. Treatment is straightforward. Affected persons are raised as female and
act female. As previously mentioned, affected persons are at increased
risk for gonadal neoplasia, and an orchiectomy is eventually needed. It
is acceptable to leave the testes in situ until after pubertal feminization, but
most surgeons would perform orchiectomies if herniorrhaphies
prove necessary before puberty. There may also be psychologic benefit
in prepubertal orchiectomies. Inguinal or intra-abdominal testes
can sometimes be removed laparoscopically.51 After orchiectomy, estrogen replacement is necessary. Because these patients
do not usually have a uterus, continuous therapy with conjugated
estrogens or other estrogen forms may be prescribed. As mentioned above, the
clinician must tailor the estrogen dosage to the individual patient, balancing
its benefits against its side effects. Vaginoplasty is
rarely necessary, but occasionally dilators may be required to increase
vaginal length. Partial Androgen Insensitivity Syndrome Partial AIS is the result of a mutation of the same androgen-receptor gene
as is involved in complete AIS.52,53 Persons with partial AIS (incomplete testicular feminization) feminize (i.e., exhibit breast development) despite having external genitalia characterized
by phallic enlargement and partial labioscrotal fusion. Both partial
and complete AIS share the following features: Bilateral testes with similar histologic findings
No müllerian derivatives
Pubertal breast development
Lack of pubertal virilization
Normal (male) plasma testosterone.
Partial AIS is an X-linked recessive condition that encompasses three entities
that once were considered separate: Lubs' syndrome, Gilbert-Drefus
syndrome, and Reifenstein syndrome. In fact, the partial AIS spectrum
even extends to include some infertile but otherwise normal males. Traditionally, diagnosis
of Reifenstein syndrome was applied to males
whose phallic development was more nearly normal than that of males
with traditionally termed incomplete androgen insensitivity. In the Reifenstein
phenotype there was no vagina-like perineal orifice, testes
were small,54 and decreased virilization was thought to be the result of inadequate
testosterone secretion. The Lubs phenotype was considered intermediate
between that of Reifenstein and that of traditionally termed incomplete
androgen insensitivity.55 This complicated stratification later proved genetically incorrect. First, males
with small testes and elevated gonadotropic levels sometimes
were found to display signs of partial AIS.56 At around the same time, Wilson and co-workers57 observed the occurrence in a single kindred of the Reifenstein phenotype
and that of partial AIS. Ten years later, Wilson and associates58 confirmed partial androgen-receptor deficiency in two persons with Lubs
phenotype. Finally, males with Reifenstein syndrome and Lubs syndrome, and
some infertile males, were observed in the same kindred.57 Thus, it was concluded that perturbation of a single gene is responsible
for all three of these disorders. The pathogenesis of partial AIS logically would appear to involve a decreased
number of receptors or qualitative defects in the androgen receptors.44,45,46,57,58,59 Complete absence of receptors has been observed, but this is more likely
to be associated with complete AIS. Surprisingly, poor correlation
exists between receptor levels (or androgen-binding affinity) and the
degree of masculinization, nor are precise correlations evident between
a specific mutation and the phenotype. Irrespective, the clinical significance
of partial AIS is that this disorder must be excluded before
the decision is made to rear a child as a male. Presence of androgen
receptors and demonstration of response to exogenous androgen is therefore
necessary to exclude the diagnosis of partial androgen insensitivity. GENETICS. Like complete AIS, partial AIS is inherited in an X-linked recessive fashion. The
chromosomal complement is 46,XY. As previously discussed, the
androgen-receptor gene has been mapped to Xq11–12. Androgen
binding studies of fibroblasts derived from the genital skin of patients
with partial AIS tend to show decreased, qualitatively abnormal, or
complete absence of androgen binding.10 Molecular analysis of the androgen-receptor gene has revealed several
different mutations in partial AIS, predictably in the androgen-binding
domains (exons 4 to 8) but sometimes in the DNA-binding domains (exons 2 and 3) (see Fig. 3). Of note is that mutations in the androgen-receptor gene have specifically
been detected in the Reifenstein phenotype,60 confirming molecularly that this phenotype is indeed the result of mutation
in the androgen-receptor gene and that partial AIS is truly an X-linked
recessive condition encompassing the entities historically considered
separate, as reasoned above. Complete deletions or point mutations resulting in premature (message) termination
are more likely to cause complete AIS than partial AIS.44 Overall, however, a molecular defect in the androgen-receptor gene is
found less often in partial AIS, suggesting that the defect may often
involve a more distal step in androgen action. Lobaccaro and colleagues41 recently studied mutations in 25 patients with partial AIS and found that
the mutations were probably the result of point mutations or microdeletions. In
addition, they were able to identify carriers in 50% of
the families with the use of exon 1 Hind III polymorphisms.41,61 DIAGNOSIS. Infants with partial AIS usually present with ambiguous genitalia. Adrenal 21-OH
deficiency can be excluded readily by chromosome studies and 17α-OH
progesterone levels.62 Ultrasound can aid in the evaluation of internal genitalia. Serum hormone
levels are assessed after hCG is administered; the presence of normal
testosterone excludes the presence of a defect in testosterone biosynthesis. An
elevated T:DHT ratio indicates 5α-reductase deficiency. If
a normal T:DHT ratio is present in an XY person with genital ambiguity, the
clinician should consider the diagnosis of partial AIS (see Fig. 5). Prenatal diagnosis for complete AIS and partial AIS is now possible
with molecular analyses of the androgen-receptor gene from trophoblastic
or amnionic DNA, if the molecular defect is known. RFLP analyses can
also be used, particularly those that utilize the polymorphic CAG repeats
in exon 1.63 MANAGEMENT. Treatment is difficult. If external genitalia are characterized by other
than simple hypospadias, a female sex of rearing is preferable. These
patients require orchiectomy and estrogen replacement (as with complete
AIS patients). If no androgen receptors are present, one must assess
the degree of androgen response in infants before considering male
sex of rearing. Patients raised as males require androgen replacement, which
may or may not be efficacious.64 Patients raised as males often require multiple urogenital reconstructive
surgeries, sometimes still with poor results.64 Furthermore, little information is available on the long-term success
of the corrective surgery and masculinization, sexual performance, and
fertility of these patients.65 Overall, it is preferable for these patients to be raised as females. |