Deficiencies of steroidogenic enzymes include a group of genetic conditions
involving adrenocorticoid and gonadosteroid biosynthesis. All are
inherited in an autosomal recessive mode. Complete or partial enzyme
deficiencies with resultant blocks in steroid synthesis result in the
accumulation of immediate precursors and a shift in steroidogenesis toward
less impeded pathways. Partial deficiencies tend to present with
a broad array of symptomatology whereas complete enzyme deficiencies are
more likely to be associated with dramatic clinical presentations. Androgen production is similar in both the adrenals and the gonads and
deficiencies may affect both targets. The testes lack 21- and 11-β-hydroxylase
and have limited aromatase activity; the major testicular
androgens are testosterone and androstenedione. The first three enzymes involved with androgen production, cholesterol-desmolase
complex, 3-β-hydroxysteroid dehydrogenase deficiency, and 17-α-hydroxylase are involved with the production of both
cortisol and testosterone and their deficiencies are typically associated
with varying degrees of adrenal insufficiency and sexual ambiguity. 17-α-hydroxylase
and 17,20 lyase are utilized only in the production
of androgens and not for cortisol synthesis (Table 2). Individuals with these deficiencies present with sexual ambiguity only. An
enzyme deficiency associated with abnormal androgen synthesis syndromes
should be suspected in 46,XY individuals with sexual ambiguity
and deficient levels of testosterone and its metabolites. TABLE 2. Abnormal Androgen Synthesis Syndromes
ACTH |
Cholesterol Desmolase Complex Deficiency (Adrenal Lipoid Hyperplasia) ↑ |
3β-Hydroxysteroid Dehydrogenase Deficiency ↑ |
17 α-Hydroxylase Deficiency ↑ |
17,20-Desmolase Deficiency N |
17 β-Hydroxysteroid Dehydrogenase Deficiency N |
Progesterone |
↓ |
↓ |
↑ |
|
|
Testosterone |
A |
A |
↓ |
↓ |
↓ |
Androstenedione |
↓ |
↓ |
↓ |
↓ |
(↑N) |
DHEA |
↓ |
↑ |
↓ |
↓ |
N |
17-OH-progesterone |
↓ |
↓ |
↓ |
↑ |
|
Aldosterone |
↓ |
↓ |
↓ |
N |
N |
Cortisol |
↓ |
↓ |
↓ |
N |
N |
17-ketosteroinds |
A |
↑ |
↓ |
↓ |
N |
17-OH-corticosteroinds |
A |
↓ |
↓ |
N |
N |
Pregnanediol |
A |
↓ |
↑ |
↑ |
|
Pregnanetriol |
A |
↓ |
↓ |
↑ |
|
A, absent; N, normal; ↑ increased; ↓ decreased.
ACTH, adrenocorticotropic hormone; DHEA, dehydroepiandrosterone.
Recent advances in molecular genetics have greatly expanded our understanding
of these disorders. Deficiencies of steroidogenetic enzymes may
result from three types of genetic aberrations: point mutations, gene
deletions or insertions, and gene conversions. Point mutations (single-base
changes) are the most common type of gene conversion. Congenital Adrenal Lipoid Hyperplasia Prader and Gurtner8 described a group of infants with severe adrenal insufficiency and massive
amounts of cholesterol deposited in the adrenal cortex and gonads
in 1955. Patients with a deficiency of cholesterol side-chain cleavage
enzyme (P-450scc) lack the ability to metabolize cholesterol in the adrenals and develop
large, yellow, foamy adrenal glands. The enzyme P-450sec initiates the steroidogenic pathway cleaving a 6-carbon side chain from
cholesterol to produce pregnenolone. This rate-limiting step is regulated
by ACTH, angiotensin II in the adrenal glands, and by gonadotropins
in the gonads. Abnormalities of this enzyme are extremely rare and
usually incompatible with life. CYP11A is the gene that encodes for cytochrome P-450scc It has been cloned and localized to the long arm of chromosome 15, it
is 20 kb long, and consists of 9 exons.9 In rabbits, homozygous CYP11A1 deletion adrenal lipoid hyperplasia consistently
occurs. However, abnormalities in the CYP11A gene have never
been observed in humans with congenital adrenal lipoid hyperplasia.10 The disorder in humans appears to be the result of perturbations of the
gene encoding, steroidogenic acute regulatory protein (StAR). The StAR
protein is responsible for delivery of precursors for cholesterol side
chain cleavage. The term acute in the name refers to the rapid response
to corticotropin stimulation to produce a 30-kd mitochondrial protein
in adrenal cells. The StAR gene is 8-kb long and consists of 7 exons
and has been mapped to 8P11.2.11 A variety of mutations have been reported.12 Approximately 100 cases have been reported and reflect preponderance among
Japanese and Koreans. At least 15 different mutations have been reported. Eighty
percent of affected Koreans and Japanese have the same
mutant allele, Gln 258 (stop). Arg 182 Leu affects 78% of Arabs
reported to have congenital adrenal lipoid hyperplasia.10 Adrenal insufficiency with low levels of cortisol, aldosterone, and androgens
is the principal manifestation. Hyperpigmentation and respiratory
distress occur in 25% of affected neonates. They present with
failure to thrive, lethargy, vomiting, diarrhea, hypernatremia, hypokalemia, and
hypertension. ACTH levels and plasma renin activity are elevated. Genetic
males present with ambiguous or female external genital. Affected
females have normal genitalia. All surviving infants have
been assigned female gender identities. Successful treatment can be accomplished
with glucocorticoids and mineralocorticoid replacement. Prophylactic
orchiectomy should be performed on affected males, and all
patients may benefit from estrogen therapy to initiate pubertal development
at the appropriate time. 3-β-Hydroxysteroid Dehydrogenase Deficiency Bongiovanni13 initially described this syndrome in 1962 as a variant of the adrenogenital
syndrome (coincident adrenal disease and abnormal genital development). The
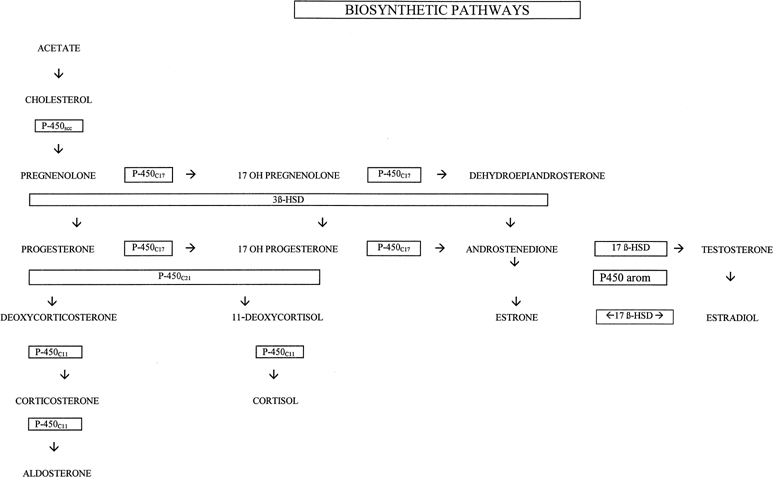
syndrome results from a block in the conversion from the Δ5 to Δ4 pathways (see 3-β-HSD in Fig. 1). Previously, two enzymes, 3-β-hydroxysteroid dehydrogenase and Δ5,Δ4 isomerase were thought to be required for this reaction, but
a single 45-kb protein has been identified with both dehydrogenase
and isomerase activities which are inseparable.14 Moreover, 3-β-hydroxysteroid dehydrogenase is the only steroidogenic
enzyme leading to the production of cortisol that does not depend
on cytochrome P-450; it requires the cofactor nicotinamide-adenine dionucleotide
oxidized (NAD+) and is a microsomal enzyme. Human 3-β-hydroxysteroid dehydrogenase (both types I and II) has
two isoenzymes and corresponding messenger RNA. These genes have been
localized to the long arm of chromosome 1 in the P11–13 band15,16 and both have four exons. Type II is expressed in gonads and adrenals. Mutations
that produce both virilization and salt wasting have essentially
abolished enzyme activity; mutations associated only with virilization
are missense mutations with 1% to 10% of normal enzyme
activity.17 The mutations producing the two phenotypes are scattered among all of
the exons. Two forms of 3-β-hydroxysteroid dehydrogenase deficiencies have
been described. The classic, as originally described by Bongiovanni, is
associated with complete block of 3-β-hydroxysteroid dehydrogenase
activity, and nonclassic, is a partial deficiency. Patients with
classic, 3-β-hydroxysteroid dehydrogenase deficiency present in
infancy with severe sodium wasting and the outcome is fatal if early
intervention is not undertaken. The adrenals and gonads are affected
with resulting deficient production of glucocorticoids, mineralocorticoids, and
sex steroids. Cortisol deficiency results in ACTH stimulation
and congenital adrenal hyperplasia. Production of both androgens and
estrogens are impaired and the major androgen produced is DHEA, which
is responsible for incomplete virilization in males and partial virilization
in females. Thus, 3-β-hydroxysteroid dehydrogenase is the
only enzyme deficiency associated with both male and female pseudohermaphroditism. Diagnosis
is established with elevated pregnenolone, DHEA, and
ratios of Δ5/Δ4 steroids. In males, testes are
usually palpable, with various degrees of hypospadias or incomplete pseudovagina
hypospadias. Females may have clitoral enlargement and partial
or complete labial fusion. The nonclassic form of 3-β-hydroxysteroid dehydrogenase deficiency
is associated with a partial deficiency and represents a less severe
clinical presentation and is much more common than the classic presentation. The
typical presentation of the nonclassic form is either premature
pubarche or pubertal hyperandrogenism.18 Clinical presentation in females may be similar to polycystic ovarian
syndrome with hirsutism, acne, menstrual irregularities, and infertility. Four
biochemical criteria have been proposed for the diagnosis of
nonclassic or late onset 3-β-hydroxysteroid dehydrogenase.19 After a 60-minute ACTH stimulation, patients should have an elevation
greater than 2 standard deviations above the mean in 17-pregnenelone, DHEA, 17-pregnenelone:17-hydroxyprogesterone ratio, and 17-pregnenelone:cortisol
ratio. Adrenal suppression with glucocorticoids is effective
treatment when the diagnosis is established. P-450c17:17-α-hydroxylase/17,20 lyase DESMOLASE DEFICIENCIES. This syndrome was initially reported by Biglieri and associates20 in 1966 in four 46,XX females and has subsequently been described in males
presenting with ambiguous genitalia.21,22 Steroid 17-α-hydroxylase/17,20-lyase (P-450c17) is a single-cytochrome-450 enzyme
that catalyzes 17-α-hydroxylase and 17,20-lyase
reactions in conjunction with NADPH-cytochrome P-450 reductase. It
is expressed in the adrenal and gonads and is required for formation
of sex hormones and glucocorticoids. Current evidence has definitively
established the existence of a single P-450 C17 enzyme with two actions.23,24 Defects in P-450c17 may be responsible for 17-hydroxylase, 17,20 lyase
deficiencies or both. A single enzyme producing both 17-α-hydroxylase
and 17,20 desmolase activity has created considerable nosologic
and genetic confusion.25 Mutations have been observed with only altered 17,20 lyase function.26 Specific regions have been identified in the rat responsible for unique
enzymatic activities.27 The human P-450c17 gene (CYP17) is a single copy gene localized to chromosome 10q24-25 with 8 exons.28,29,30 It contains 508 amino acids and has a molecular weight of approximately 57 kd. The
gene has been completely analyzed and sequenced. Mutations
have been found in the structural gene and regulatory sequences determining
expression of gene CYP17. A number of missense mutations have
been described usually ablating P-450c17 function. P-450c17 is expressed in the adrenal gland and the gonads; deficiencies
lead to inadequate cortisol and sex steroid production. ACTH is increased
by the cortisol deficiency and the block at 17-α-hydroxylase
promotes excessive production of 17-deoxysteroids, especially deoxycorticosterone (DOC), which has potent mineralocorticoid activity. Elevated
mineralo-corticoid levels produce hypernatremia with low renin hypertension, hypokalemia, and metabolic alkalosis. Deficient sex steroids
result in elevated gonadotropin levels. Clinical presentation in males includes sexual ambiguity, which correlates
with the severity of the block in P-450c17 activity and the resulting
impairment in testosterone production. Testes may be intra-abdominal
within the inguinal canal or within the labioscrotal folds. Inguinal
hernias are a common accompaniment. Males typically have hypoplastic
wolffian structures and females have normal but infantile müllerian
structures. Most patients present at puberty with primary amenorrhea
or delayed pubertal development.31 A few patients have been diagnosed in infancy, presenting with ambiguous
genitalia and hypertension. Affected females are more likely to be
hypertensive while 17-α-hydroxylase deficient males are more often
normotensive. Relevant laboratory findings include high serum levels of 17-deoxysteroids, progesterone, DOC, and corticosterone. Serum levels of 17-hydroxysteroids, including
cortisol and DHEA, testosterone, and estrogens are
undetectable. P-450c17 deficiency is treated with glucocorticoid suppression
similar to therapy in patients with congenital adrenal hyperplasia. Hormone
replacement therapy may be given at puberty to initiate
secondary sexual development. Individuals with ambiguous genitalia undergo
sex assignment at birth and may be raised as males if phallic development
is adequate or may undergo phallic reduction and gonadectomy
if not. 17-β-Hydroxysteroid Dehydrogenase Deficiency (17-Ketosteroid Reductase) A defect in the last step of testosterone synthesis, the conversion on
androstenedione to testosterone (see 17-β-HSD in Fig. 1), was first described by Neher and Khant in 1965.32 Several additional males, including siblings, have been described subsequently, having
less compromise of virilization than those with other
causes of impaired androgen synthesis.33,34,35,36,37,8,39 Some affected individuals are born and reared as females but undergo marked
virilization at puberty, similar to men with 5-α-reductase
deficiency. The disorder is rare but relatively common in certain populations
such as among Arabs living in the Gaza strip.40 17-β-HSD is an NADPH-dependent microsomal enzyme that catalyzes
the reversible reduction necessary to form testosterone from estrone. Five
isozymes have been cloned that catalyze the oxydoreduction of androstenedione, testosterone, and DHT, estrone, and estradiol.41 The 17-β-HSD gene has been localized to chromosome 9. At least 14 mutations
in the 17-β-HSD enzyme gene have been identified from
different ethnic groups. Type 3 17-β-HSD isozyme consists of 11 exons
and has its primary action in the gonads. Deficiency of this
enzyme is associated with the form of male pseudohermaphroditism most
frequently seen in Gaza. The most common mutation is a missense mutation
in exon 3 that reduces enzyme activity to 15% to 20% of
normal.42 Other mutations reported in additional populations involve all 11 exons
and include frame shifts, splice junction alterations in addition to
the most common missense mutations. Males affected with the 17-β-HSD gene have normal wolffian structures
but ambiguous genitalia. Phenotypic variability has been reported
within families with the same homozygous mutation.43 Many have been raised as females but virilized at puberty and subsequently
adopted a male gender role. Some develop gynecomastia. These individuals
have elevated serum levels of androstenedione resulting from an
inability to make testosterone, increased DHEA because of an inability
to make Δ5 androstenediol, and high estrone levels resulting
from an inability to convert to estradiol. At puberty, testosterone and
estradiol levels are markedly decreased over normal values. Diagnosis
is established by endocrine evaluation and mutation analysis.43 Treatment involves gonadectomy and estrogen supplementation at puberty
for those raised as females. One individual with teratocarcinoma and metastasis
has been reported.44 Individuals raised as males benefit from repair of the hypospadias and
may require testosterone replacement. These individuals do not achieve
spermatogenesis, and histologic examination in the postpubertal male
reveals Leydig cell hypoplasia. Leydig Cell Hypoplasia Patients with Leydig cell hypoplasia present with a decreased response
of the Leydig cells to luteinizing hormone. Patients with this syndrome
may present with variable findings ranging from frankly ambiguous genitalia
to normal male genitalia with micropenis. A recent study of three
affected brothers with a mild form of Leydig cell hypoplasia revealed
a homozygous missense mutation resulting in a substitution of a lysine
residue for an isoleucine residue at position 625 of the receptor.45 Studies showed a correlation between the severity of clinical presentation
and overall receptor signal capacity, a combination of cell-surface
expression and coupling efficiency. Instability of Multiple P-450 Enzymes A 6-month old, 46,XY infant was described with genital ambiguity and multiple
enzyme defects, including 17-α-dehydroxylase, 21-hydroxylase, and 17,20-desmolase
in 1985. Peterson and associates46 suggested a new form of male pseudohermaphroditism, postulating that multiple
adrenal biosynthetic abnormalities arose as a consequence of an
abnormality of the cytochrome P-450 system. Enzymes involved in adrenal
biosynthesis include both mitochondrial and cytoplasmic P-450 enzymes.47 Smith-Lemli-Opitz Syndrome Smith-Lemli-Opitz Syndrome (SLO) is a relatively common (1:10,000) autosomal
recessive syndrome affecting 46,XY individuals with variable expression
ranging from hypospadias to male pseudohermaphrodism.48 Of the two types described, hypospadias is the most common abnormality
identified with type I SLO. Type II SLO has been associated with ambiguous
external genitalia and even frank sex reversal.49 A mutation of the gene whose enzyme converts 17-hydroxycholesteral to
cholesterol has been implicated in type I and type II.50,51 The most common associated molecular abnormality is an exon-intronic splicing
defect. Maternal serum estriol levels are low to nondetectable
in pregnancy. Dehydropregnanetriol and dehydro-estriol are detectable
in maternal serum and urine during affected pregnancies. These compounds
are not detectable in normal pregnancies, making antenatal testing
for SLO feasible, and treatment with a high cholesterol diet has been
considered.52 Deficiency of Steroidogenic Factor-1 Steroidogenic factor-1 (SF-1) is an orphan nuclear receptor encoded by
the FTZ1 gene located on 9q33 and translated into a zinc finger protein. It
is considered to be an orphan receptor because no ligand has yet
been identified. Prior to identification of human cases, disruption of
FTZ1 in mice (knockout) was observed to disrupt adrenal and gonadal
development as well as hypothalamic and pituitary gonadotrophic tissue.53,54 On the basis of the mice studies, it was predicted that mutations in human
FTZ1 would produce abnormal SF-1 receptors and lead to significant
abnormalities of sexual development. Achermann55 described the first human case of SF-1 deficiency caused by mutant FTZ-1. The
affected 46,XY individual demonstrated primary adrenal failure, female
genitalia, streak gonads, and normal müllerian-derivative
responsive hormones. After appropriate studies were completed to exclude
congenital adrenal lipoid hypoplasia, the proband was demonstrated
to be heterozygous for a 2bp substitution in FTZ-1 at codon 35. SF-1 is
believed to regulate repression of AMH. The presence of normal müllerian
structures in the reported patient despite genital and
gonadal abnormalities supports such a role for SF-1. |