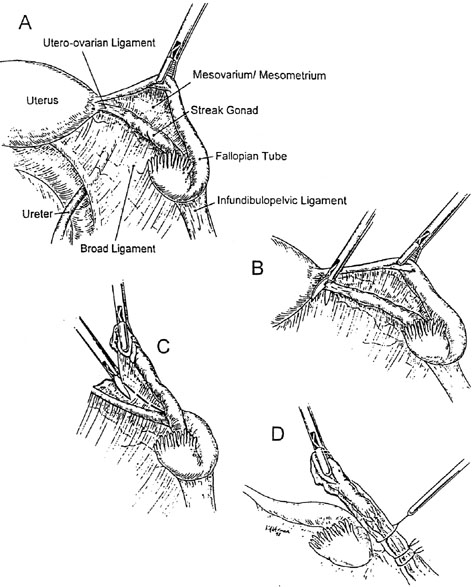
Primordial germ cells originate in the endoderm of the yolk sac and migrate to the genital ridge to form the indifferent gonad. 46,XY and 46,XX gonads are initially indistinguishable. Indifferent gonads develop into testes if the embryo, or more specifically the gonadal stroma, is 46,XY (Fig. 1). This process begins approximately 43 days after conception. Testes become morphologically identifiable 7 to 8 weeks after conception (9 to 10 gestational or menstrual weeks).
Sertoli cells are the first cells to become recognizable in testicular differentiation. These cells organize the surrounding cells into tubules. Both Leydig cells4 and Sertoli cells5 function in dissociation from testicular morphogenesis, consistent with these cells directing gonadal development rather than the converse. These two cells secrete hormones that direct subsequent male differentiation (see Fig. 1).
|
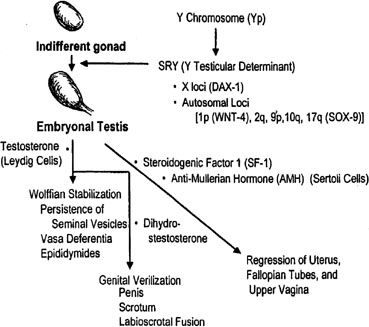
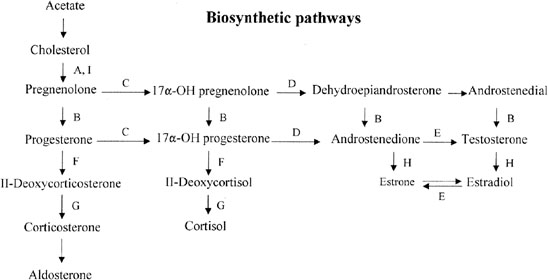
Fetal Leydig cells produce an androgen, testosterone, that stabilizes wolffian ducts and permits differentiation of vasa deferentia, epididymides, and seminal vesicles. After conversion by 5α-reductase to dihydrotestosterone (DHT), external genitalia are virilized. These actions can be mimicked by the administration of testosterone to female or castrated male embryos, as demonstrated clinically by existence of teratogenic forms of female pseudohermaphroditism.
Fetal Sertoli cells produce anti-müllerian hormone (AMH), also known as müllerian inhibitory substance (MIS). This glycoprotein diffuses locally to cause regression of müllerian derivatives (uterus and fallopian tubes). When AMH is chronically expressed in XX transgenic mice, oocytes fail to persist, tubule-like structures develop in gonads, and müllerian differentiation is abnormal.6 Thus, AMH may have functions related to gonadal development as well. Steroidogenic factor 1 (Sf-1) appear to regulate AMH, given that a uterus was present in a gender-reversed XY female lacking SF-1.7
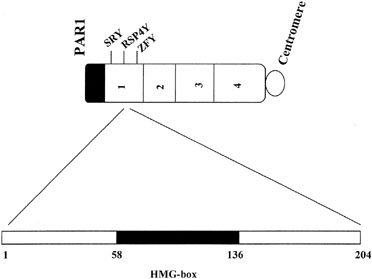
In the absence of a Y chromosome, the indifferent gonad develops into an ovary. Transformation into fetal ovaries begins at 50 to 55 days of embryonic development. Whether female (ovarian) differentiation is truly a default (constitutive) pathway or whether a yet to be determined gene product directs primary ovarian differentiation is uncertain. That 46,XY individuals with certain syndromes (Gardner-Silengo-Wachtel) may show oocytes, as may XY gonadal dysgenesis neonates,8 favors the default hypothesis. Partial ovarian function was also found in a gender-reversed XY case having adenovo mutation (Gln2Stop) at the 5[prime] end of SRY.9 Relevant is that germ cells are present in 45,X embryos,10 only to undergo atresia at a rate more rapid than that occurring in normal 46,XX embryos; thus, pathogenesis involves not only failure of germ cell formation but increased atresia. Perhaps the same phenomenon occurs in XY gonadal dysgenesis.
Internal ductal and external genital development is secondary to but independent of gonadal differentiation. In the absence of testosterone and AMH, external genitalia develop in female fashion. Müllerian ducts form the uterus and fallopian tubes, and wolffian ducts regress. This scenario occurs in normal XX embryos as well as in XY embryos (animals) castrated before testicular differentiation.