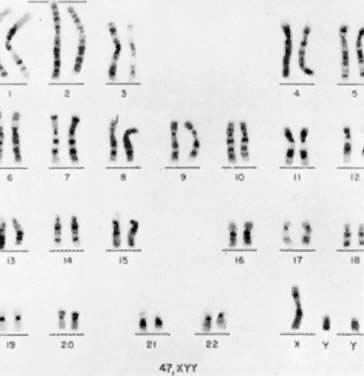
47,XYY
FREQUENCY.
The first individual with a 47,XYY chromosomal complement (Fig. 1) was described by Sandberg and co-workers in 1961.2 For several years thereafter, relatively few cases were reported, but in 1965-1966, several investigators reported that among mentally retarded criminals the prevalence of 47,XYY was higher than expected by chance, creating a resurgence of interest in this disorder.3,4,5,6 Jacobs7 summarized the data collected from eight chromosome surveys of unselected infants.8,9,10,11,12,13,14,15 Among 39,557 males studied, 38 (0.1%) were found to have 47,XYY complements. These data correspond closely to those of two large sex chromatin surveys of newborns16,17 as well as recent data reported by Hecht and Hecht18 demonstrating the frequency of XYY in newborn males to be 1 in 975.
|
Among a total of 13,406 male infants studies, 9 (0.07%) were determined to have two Y-bodies. Subsequently, 47,XYY karyotypes were confirmed in these 9 infants. There has been some suggestion that the incidence of 47,XYY males may be lower among blacks than among whites.19
CELLULAR ETIOLOGY.
The most likely origin of 47,XYY is paternal nondisjunction at meiosis II, resulting in 24,YY spermatozoa, followed by syngamy with a normal 23,X ovum. A 24,YY spermatozoon would also be a normal product of meiosis in a male with a 47,XYY chromosomal constitution; however, this phenomenon probably accounts for relatively few 47,XYY men.20 Alternatively, postzygotic nondisjunction in a 46,XY zygote could give rise to 45,X/47,XYY mosaicism. Either exclusion of the 45,X line or selection in favor of the 47,XYY line could result in an apparently “pure” 47,XYY zygote. The occurrence of double aneuploidy, for example, 48,XYY,+21, raises the possibility that genetic factors could be important in the etiology of polysomy Y.21 Finally, parental age appears to play no important part in the genesis of 47,XYY.22
CLINICAL FINDINGS.
Psychologic and Intellectual Function.
The first psychologic studies of 47,XYY males were those of Price and co-workers,23,24 who studied inmates at a Scottish maximum security prison. Compared with 18 46,XY inmates, 9 47,XYY inmates (1) incurred their first conviction at a younger age (mean age 13.1 years for 47,XYY; 18 years for 46,XY, (2) less often had a sibling who had received a conviction (1/31 siblings of 47,XYY inmates; 13/63 sibs of 46,XY inmates), and (3) committed crimes against property more often than crimes against persons. These observations suggested that 47,XYY inmates are incarcerated for different reasons than are other inmates. Probably the most objective study was from Denmark, conducted by Witkin and co-workers,25 in which a 42% (5/12) rate of criminality was found among 47,XYY males compared with 9.3% in 4096 46,XY controls. The difference is about 4.5-fold and is significant at the 0.05 level. Hook26 suggested that despite the observation that intelligence quotients of 47,XYY males are lower than 46,XY controls in exclusively penal settings, it is not the lower intelligence per se that is exclusively responsible for the higher frequency of incarceration but rather the significant increase in risk of social maldevelopment associated with the 47,XYY genotype.
On the other hand, some investigators object to these conclusions regarding the increased frequency of social maldevelopment and predisposition to “criminality.”27 Accurate assessment of the intellectual and psychologic problems in 47,XYY individuals therefore awaits the results of larger prospective surveys of neonates or randomly ascertained adults. Robinson and associates28 summarized the findings of 43 47,XYY infants prospectively ascertained through various chromosome surveys of unselected neonates. About a third of the children showed delayed speech or language development (7 of 18), an increased frequency over their siblings and controls. IQ scores ranged from 78 to 145; however, there was a slight skew to the left in IQ distribution, with 14 of 37 children being in the 70 to 89 IQ range. Gross motor development was generally found to be normal, although there was a suggestion of fine motor problems. There currently appears to be insufficient information to make any conclusions regarding the frequency or magnitude of the increased risk for social pathology28,29 in 47,XYY individuals.
Somatic Abnormalities.
In summarizing the data concerning the 43 47,XYY neonates who were prospectively ascertained through newborn surveys, Robinson and associates28 found that birthweights, length, head circumference, and course during the first year of life were within normal ranges. There was no clear “XYY syndrome” identifiable at birth, with the majority of neonates being completely normal in appearance. There was only one major anomaly, that is, congenital hip dislocation. Eight neonates had one or more minor anomalies, including clinodactyly (2 cases) with a single crease of the fifth finger in one, inguinal hernia (2 cases), abnormal ears (2 cases), pectus carniatum, borderline large head, asymmetric head, strabismus, epicanthic folds, micrognathia, acne, philosis, simian crease, and an undefined heart murmur.
In addition to the aforementioned clinical manifestations, 47,XYY individuals are sometimes quite tall. In the Scottish prison surveys, 47,XYY inmates30 had a mean height of 71.3 inches (181.2 cm) compared with 67.2 inches (170.7 cm) for 46,XY inmates. Some investigators believe that electrocardiographic abnormalities are present31,32; however, others have not been able to confirm such abnormalities.33,34 Dermatoglyphic analyses have shown decreased total digital ridge counts consistent with the general observation of an inverse relationship between sex chromosome polysomy and total ridge count.35,36 The sizes of deciduous teeth of 47,XXY individuals have been found to be larger than controls, suggesting that the Y chromosome regulates quantitative variation of dental growth.37 Finally, abnormal electroencephalograms have been reported in 47,XYY men in psychiatric hospitals and prisons38; however, persons resident in institutions are more likely to exhibit abnormal electroencephalograms compared with normal individuals.
The evidence of fertility of 47,XYY males is limited; however, the sons of 47,XYY males usually show normal 46,XY complements.20,37,38,39,40,41 Seminiferous tubules characterized by spermatogenic arrest are detected in about 50% of 47,XYY males, and about 30% of tubules consist solely of Sertoli cells.42 Sperm counts may be low, even if testes are normal in size.42 Although somewhat controversial, the consensus is that testosterone levels are within normal ranges in most 47,XYY males. Migeon and associates43 investigated the plasma testosterone levels in 15 47,XYY males from the United States and 15 such individuals from England, whose ages ranged from 20 to 50 years. Seventeen of these 30 individuals showed a normal range of testosterone. Seven showed a level over 2 standard deviations above the mean and 6 showed a level under 2 standard deviations below the mean (normal range, 460 1 mμg/100 ml; SD, ± 125 mμg/100 ml).
GENETIC COUNSELING.
Theoretically, meiosis in 47,XYY males should lead to 50% normal gametes with a single X or Y and to 50% gametes with 24,YY or 24,XY complements; hence, 50% of the offspring should be trisomic, either 47,XYY or 47,XXY. However, offspring of 47,XYY males have usually been reported to be chromosomally normal. Some 47,XYY men have purportedly had offspring with trisomy 21,44 which raises the possibility that the abnormal sex chromosome constitution affects segregation of autosomal chromosomes as well. Further investigations are needed to understand gametogenesis in 47,XYY individuals. Until such data become available, it seems warranted to offer prenatal diagnosis in this situation.
If fetal cytogenetic studies reveal a 47,XYY complement, the couple must be fully informed regarding potential psychiatric, social, and somatic abnormalities of 47,XYY individuals. As in all cases of prenatal diagnosis, the couple must make the ultimate decision of whether to continue the pregnancy. Their decision must be supported by the physician and the entire genetic counseling team.
47,XYY and Female Phenotype
Individuals with 47,XYY cells may occasionally have female or ambiguous external genitalia and may be categorized into three groups:
- 45,X/47,XYY mosaicism. These individuals might be expected to display a
wide clinical spectrum, that is, (1) almost normal male development,45 (2) ambiguous external genitalia associated either with two dysgenetic
testes or with a unilateral streak gonad and a contralateral testis (mixed
gonadal dysgenesis), or (3) female external genitalia and bilateral
streak gonads.46 Mosaicism of the 45,X/47,XYY type has been detected in two and possibly
three siblings whose parents were consanguineous47,48; therefore, in some cases this mosaicism may be influenced by genetic
factors.
- Suspected 45,X/47,XYY mosaicism, 47,XYY cells may be the only demonstrable
cells in individuals who have both müllerian derivatives and
genital ambiguity. The presence of müllerian derivatives suggests
mosaicism.
- 47,XYY individuals with female or ambiguous external genitalia but no uterus. The
absence of a uterus suggests a disorder other than undetected
mosaicism because a uterus is usually present in 45,X/47,XYY individuals. Some
cases may represent the coincidental association of testicular
feminization and 47,XYY; others are more difficult to explain and
could be teratogenic in etiology.
48,XYYY
Relatively few cases of 48,XYYY have been reported.49,50,51,52,53 Presumably 48,XYYY arises from Y nondisjunction in mitosis of spermatocyte formation followed by a Y nondisjunction during meiosis I and subsequent chromatid nondisjunction of one Y during meiosis II, resulting in a sperm bearing three Y chromosomes.
Clinical features which appear similar in some 48,XYYY males include low normal intelligence quotient, behavioral problems with aggressive outbursts, repeated pulmonary infections during childhood, sparse body hair, clinodactyly of fifth fingers, acne, and hypotrophic testes.50,52 However, Hunter and Quaife51 described a patient exhibiting no stigmata other than sterility.
48,XXYY and 49,XXXYY
48,XXYY and 49,XXXYY are associated with Klinefelter's syndrome phenotype (see related chapter in this volume).