Attempts to categorize leiomyomas further have been aided by cytogenetic
analyses of resected samples. Approximately 25% to 40% of fibroids
show karyotypically detectable chromosomal abnormalities that are both
nonrandom and tumor-specific.10,11
These samples have been classified into several cytogenetic categories
based on the chromosome aberration present and include the following subgroups:
t(12;14)(q14-q15;q23–24), del(7)(q22q32), rearrangements involving
6p21, 10q, trisomy 12, and deletions of 3q. Although no relationship
between patient age or parity and the type of chromosomal abnormality
has been identified, a recent study positively correlated the presence
of a cytogenetic abnormality with the anatomic location of a uterine fibroid.12
In this study, submucous fibroids were consistently shown to have fewer
cytogenetic abnormalities (12%) compared with intramural (35%)
and subserous (29%) fibroids. These data complement earlier work correlating
increased tumor size with presence or type of chromosomal abnormalities.10,13,14
Clonality Studies of Uterine Leiomyomas
Cytogenetic analyses of multiple fibroids from a single uterus have demonstrated that the tumors can harbor different chromosomal changes, suggesting that each fibroid develops independently. X-inactivation studies, which exploit the phenomenon of Lyonization, have clearly demonstrated that fibroids develop as clonal lesions.15,16,17,18 Initially, X-linked glucose-6-phosphate dehydrogenase (G6PD) isoenzyme analysis was used to demonstrate the independent clonal origin of multiple tumors within a single uterus.17,18 However, a low degree of G6PD isoenzyme polymorphism among most females across various ethnic groups limited the usefulness of this analysis. Another approach, based on the CAG repeat polymorphism in the X-linked androgen receptor gene, was used in a more recent study to examine clonality.15 Experiments in this study demonstrated a random pattern of inactivation among multiple tumors: individual tumors exclusively expressed one or the other allele, confirming the hypothesis that multiple tumors within a single uterus arise independently. Another study, using DNA digestion with methylation-sensitive restriction enzymes to examine differences in methylation between the active and inactive X chromosome at the androgen receptor locus, confirmed the monoclonal nature of fibroids.19 It is possible, however, that some leiomyomas develop from a common precursor cell; the discovery of identical cytogenetic abnormalities in multiple fibroids from the same patient argues for this path of development.10 Yet these changes may simply be representative of neoplastic smooth muscle. A study of a single patient with two independent fibroids, each showing different patterns of X-chromosome inactivation but identical del(7)(q21.2q31.2) derivative chromosomes, certainly supports this latter hypothesis.20
Studies of tumors that initially appeared to be polyclonal have shown that two independent fibroids can actually develop in extremely close proximity to each other.21 The discovery of heterogeneity of chromosomal aberrations is consistent with the multistep hypothesis of tumor development, in which the function (or dysfunction) of several genes at multiple loci results in fibroid growth. Abnormalities at several loci have been documented in individual tumors, and this heterogeneity may explain clinicopathologic differences seen in fibroids, including variation in size or response to hormonal treatments. With such heterogeneity it is possible that, rather than a required order to the mutation of critical genes, the combined action of individual genes and accumulation of mutated loci contribute to the growth of leiomyomas. This phenomenon has been studied and described in detail for a number of malignant tumors.22,23
The t(12;14) Subgroup
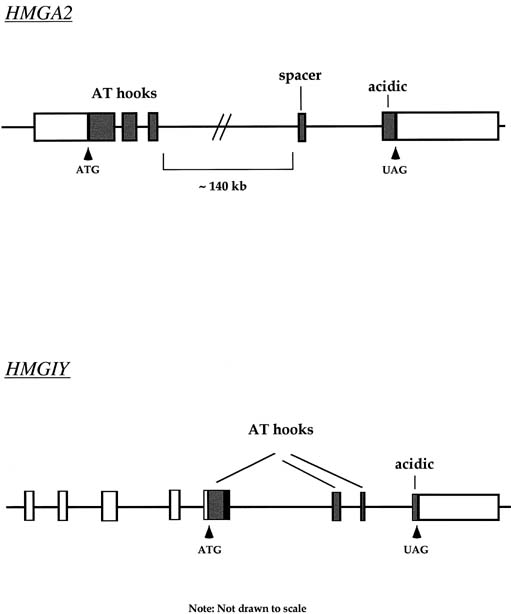
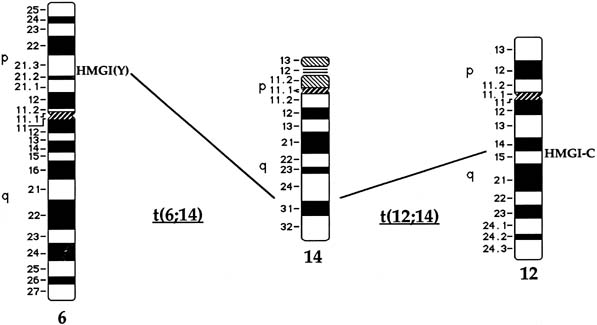
Approximately 20% of karyotypically abnormal leiomyomas show a characteristic t(12;14) rearrangement, the most commonly seen chromosomal translocation in fibroids as well as the first cytogenetic anomaly to be associated specifically with uterine leiomyomas.24 The finding of rearrangements involving the same region of 12q in other mesenchymal solid tumors (e.g., angiomyxomas, breast fibroadenomas, endometrial polyps, hemangiopericytomas, lipomas, pulmonary chondroid hamartomas, salivary gland adenomas) supported the notion that a critical gene(s) for the process of tumorigenesis mapped to 12q14–15 (Fig. 1).25,26,27,28,29,30,31,32,33 The fact that t(12;14) was frequently the sole detectable chromosomal alteration in a subset of fibroids lent additional support to the importance of genes mapping to the breakpoints involved in this translocation. After many efforts to map the critical region on chromosome 12, a single yeast artificial chromosome (YAC) clone was found to span cytogenetic breakpoints in uterine leiomyoma, pulmonary chondroid hamartoma, and lipoma specimens with t(12;14) translocations, and therefore was likely to include the critical gene.34 The HMGA2 [formerly designated as HMGI(C)] gene was first considered a positional candidate because it mapped within this YAC clone and eventually was identified as the disrupted gene in a series of mesenchymal tumors.34,35 A candidate gene on chromosome 14 that may be involved in t(12;14) is RAD51L1, which was recently identified as a partner of HMGA2.36,37RAD51L1 is a member of the recA/RAD51 recombination repair gene family. However, it is important to note that RAD51L1 spans a large genomic region of approximately 700 kilobits that potentially contains multiple other genes that may play a role in the pathobiology of fibroids.
|
The del(7)(q22q32) Subgroup
Del(7)(q22q32), an interstitial deletion of chromosome 7 involving bands q22-q32, is a common chromosome abnormality of uterine leiomyomas, with an observed frequency of approximately 17% in karyotypically abnormal fibroids.38,39,40 Loss of genetic material from 7q and rearrangements specifically involving band q22 have been found more consistently in uterine leiomyomas than in any other solid tumor. The finding of this particular anomaly as the sole alteration in some fibroids indicates its possible role as an early genetic event. Interestingly, del(7) can be observed simultaneously with t(12;14), suggesting involvement of del(7) in the karyotypic evolution of fibroids, although the t(12;14) can be similarly observed as an isolated aberration.41 Chromosome 7 rearrangements other than deletions have also been documented in fibroids and include various translocations, implicating 7q22 as the pathogenetic region.39
As is often the case in the identification of pathogenetic chromosomal segments, cytogenetic translocations involving chromosome 7 have been instrumental in narrowing the location of critical genes (see Fig. 1C). Early studies reported deletions of chromosome 7q from band q11.23 to q36.42,43,44 With regard to mapping disease genes, this region, on the order of 2 megabases of DNA, represented an inordinately large distance in which to search. Further work limited the minimal region to band q22, and an additional study reporting three leiomyomas with translocations involving 7q22 confirmed the importance of this region to leiomyomas.38,39,45 Interestingly, one report found a subset of tumors with two discrete deletions mapping to the long arm of chromosome 7.46
It is notable that cultured fibroid specimens with chromosome 7 deletions or translocations are usually found in the mosaic state with 46,XX karyotypically normal cells.47 These cultures appear to be highly unstable and frequently lose the chromosomally aberrant line. Interestingly, cells with the del(7)(q22q32) appear to be more stable in culture when the t(12;14) is also present. The instability of del(7)(q22q32) cells without chromosome 12 aberrations is intriguing in light of the observation that fibroids with chromosome 12 abnormalities or rearrangements are often larger than tumors with chromosome 7 abnormalities, and larger fibroids are more likely to be chromosomally abnormal than smaller ones.14 Observations of tumor instability in culture and decreased tumor size in association with the del(7)(q22q32) suggest that a tumor suppressor gene at 7q22 may be regulating cellular growth.
Interstitial deletions of the long arm of chromosome 7, as well as translocations involving the same region, have been reported in other benign mesenchymal tumors, including lipomas and endometrial polyps.29,48,49 In certain instances, the chromosome 7 abnormality has been the only anomaly present in the tumor. Interestingly, 7q deletions have also been documented in a subset of patients with primary acute nonlymphocytic leukemia (7.6%), with or without subsequent chemotherapy, and in those with myelodysplastic syndrome (19%). These specific deletions, considered a poor prognostic factor for certain hematological disorders/neoplasias, are observed with even greater frequency in patients with secondary acute myelocytic leukemia (26.8%) and secondary myelodysplastic syndrome (41%).42,50,51,52 In one study, fluorescence in situ hybridization (FISH) analysis with markers in the 7q21-q31 region was used to study an established cell line from a leukemia patient and to limit the critical region in acute nonlymphocytic leukemia to a 2-megabase interval.53 Patients with this particular cytogenetic abnormality are more likely to experience remissions of limited duration and have disease refractory to therapy. In sharp contrast, 7q deletions in benign mesenchymal tumors appear to have no adverse clinical significance.
Dissection of 7q22 to study leiomyoma-specific sequences is complicated by the fact that this is a gene-dense region including genes involved in developmental processes (DLX5, DLX6) and collagen metabolism (collagen type 1, procollagen C-endopeptidase enhancer), as well as those that encode acetylcholinesterase, plasminogen activator inhibitor type 1, and mucin.54,55 A potential tumor-suppressor gene, CUTL1, which encodes a homeodomain protein, has been mapped to 7q22, and loss of heterozygosity (7 of 50 samples) or reduced expression (8 of 13 samples) has been demonstrated in a subset of leiomyomas.56 The DNA-replication initiation factor ORC5L has also been mapped to 7q22 and is deleted in a subset of leiomyomas.57 Because ORC5 protein is a subunit of the DNA replication complex so intimately connected to cell growth, it is tempting to speculate that loss of this protein may somehow contribute to instability and poor viability of leiomyomas with 7q22 deletions. Despite identification of several intriguing positional candidate genes, to date none has been proved to have a causative role in the genesis of leiomyomas. Clearly, because the region is both physically large and gene-rich, identification of pathogenetic sequences is a task of considerable magnitude. Most recently, molecular analysis of band 7q22 in uterine leiomyomas has defined a 10-centiMorgan critical region common to tumors with cytogenetic deletions.58 Taken together, these findings indicate an important role for a genetic locus at 7q22 in the development of uterine leiomyomas. Although the discovery of tumors in which the sole documented cytogenetic abnormality is del(7)(q22q32) may support an early causative role for this mutation, it is clear that additional submicroscopic mutations affecting other loci cannot yet be excluded.
Rearrangements of Chromosome 6 in Band p21
Rearrangements of band 6p21 have been observed frequently in the same group of previously mentioned mesenchymal tumors, including lipomas, pulmonary chondroid hamartomas, endometrial polyps, and uterine leiomyomas (see Fig. 1).59,60,61,62 In fibroids, these rearrangements occur with a frequency of less than 5% and include t(6;14), t(6;10), and t(4;6), as well as inversions and translocations involving other chromosomal partners. After the architectural factor HMGA2 was identified as a gene involved in the pathobiology of this group of benign tumors, and given the observed rearrangements involving 6p21 in a subset of tumors, a high-mobility group (HMG) family member, HMGA1 [formerly designated as HMGIY], became an attractive positional candidate gene for fibroids. A single genomic clone including HMGA1 was found to span the breakpoint in a uterine leiomyoma with a complex rearrangement involving 6p21.3.63 Subsequent studies demonstrated rearrangement of the HMGA1 locus in hamartoma of the breast,61 a pericentric inversion of chromosome 6 involving band p21 in a uterine leiomyoma,60 and a translocation leading to an intergenic fusion with the LAMA4 gene (laminin α4) in a pulmonary chondroid hamartoma.59