Adrenal Gland Physiology There are a few studies on the size of the adrenal glands during pregnancy, but
there is no conclusive evidence of any significant change in
weight.128 Histologically, the adrenal gland is divided into three zones: zona glomerulosa, zona
fasciculata, and zona reticularis. The glomerular zone
produces aldosterone; the middle, or fascicular, zone produces mainly
cortisol; and the inner, or reticular, zone produces androgens and a
minimum amount of estrogen. Cortisol is the chief secretory product of human adrenal glands. In addition
to having glucocorticoid activity, it has a potent anti-inflammatory
effect when used in pharmacologic doses. Cortisol is present in the
circulation in at least three forms: bound to a specific binding protein (corticosteroid-binding globulin [CBG] or transcortin), bound
to albumin, and in the free form. In the nonpregnant woman, 70% to 80% is
bound to CBG, and only 10% to 15% is in the free or unbound
fraction. In pregnancy, there is an increase in the plasma levels of
CBG, and by the end of the third trimester the serum levels of CBG have
doubled. At the same time, there is an increase in the free or unbound
cortisol. This is the only biologically active fraction of cortisol, and
its elevation is demonstrated by the first trimester of gestation.129,130 Aldosterone is the most potent mineralocorticoid; its daily secretion rate
in nonpregnant women is about 50 to 250 μg on a normal sodium intake. Its
secretion is controlled by the renin - angiotensin system, by
potassium, and, to a lesser degree, by ACTH. Aldosterone secretion
rate and plasma concentration increase during pregnancy, with the first
changes occurring as early as 2 weeks after conception.131 The adrenal cortex secretes dehydroepiandrosterone (DHEA) and dehydroepiandrosterone
sulfate (DHEAS). The DHEA production rate is increased during
pregnancy. It is aromatized to estradiol and estrone by the term
placenta and may contribute up to 9% of circulating estradiol.132 Cushing's Syndrome Cushing's syndrome is defined as a constellation of signs and symptoms
due to chronic excessive production of glucocorticoids by the adrenal
glands. As in many other endocrine conditions, women between the
ages of 20 and 40 years are more affected than men, with a ratio of about 9:1. Cushing's
syndrome can be classified as ACTH dependent or
ACTH nondependent (Table 6). ACTH-dependent Cushing's syndrome is caused by hyperproduction
of glucocorticoids by the adrenal glands secondary to excessive or inappropriate
secretion of ACTH or ACTH-releasing hormone by an ectopic tumor. ACTH-nondependent
Cushing's syndrome is caused by an intrinsic
disorder of the adrenal gland, such as a benign or malignant adrenal
tumor. Alternatively, it may be iatrogenic, secondary to pharmacologic
doses of glucocorticoids in the treatment of systemic disease. In
women of childbearing age, the most common cause of Cushing's syndrome
is bilateral adrenal hyperplasia, which accounts for 75% of all
cases. Adrenal tumors are unilateral, are usually benign (adenoma), and
represent 20% of all cases. In pregnancy, however, adrenal adenomas
are as common as bilateral hyperplasia in the etiology of Cushing's
syndrome. Adrenal carcinoma is rare, although several cases have been
reported during pregnancy. Ectopic ACTH syndrome is unusual in young
people, but occasional cases have been reported in pregnancy.133,134,135,136,137,138,139 TABLE 6. Classification of Cushing's Syndrome
ACTH dependent Chronic administration of ACTH
Excessive production of pituitary-ACTH (Cushing's disease) (bilateral
adrenal hyperplasia)
Ectopic ACTH
Non-ACTH dependent Chronic administration of synthetic corticosteroids
Adrenal adenoma
Adrenal carcinoma
ACTH, adrenocorticotropic hormone
Cushing's syndrome is characterized clinically by a slow progression
of symptoms. These manifest as muscle weakness, personality changes, oligomenorrhea
or amenorrhea, hirsutism, weight change, and back pain
due to osteoporosis. One of the first common manifestations in women
is oligomenorrhea, which progresses to amenorrhea. Hence, pregnancy in
women with Cushing's syndrome is unusual. The anovulation is due
to the suppression of pituitary gonadotropin by high levels of glucocorticoids. An
increased incidence of thromboembolism also has been reported. On
physical examination, hypertension, truncal obesity, fat deposition
in the supraclavicular areas and upper dorsal spine (buffalo
hump), moon face, atrophy of skin that is easily bruised, and purple striae (over
abdomen, axillae, and hip areas) are common clinical features. Weakness
due to muscular atrophy can be very marked, particularly
in the quadriceps muscles; the patient may be unable to rise from a deep
knee bend without assistance. Headaches and visual field disturbances
due to a large pituitary tumor are rare. Hirsutism, sometimes of the
lanugo type, is common. The severity of virilization depends on the
amount of androgen produced; in patients with adrenal adenoma the secretion
of androgens is low, whereas it is moderately high in patients
with bilateral adrenal hyperplasia. Masculinization is rare, and when
it occurs it is due to an adrenal carcinoma producing large amounts of
androgens. Kidney stones due to hypercalciuria are reported to occur
in 15% to 20% of patients. An increase in the hematocrit, leukocytosis with relative lymphopenia, and
eosinopenia are usually seen. An abnormal glucose tolerance test has
been reported in over 50% of patients, and frank diabetes has been
seen in 20% to 30%. Mild hypokalemia with metabolic alkalosis is not uncommon. Radiologic
examination of the chest may show an enlarged mediastinum
secondary to fat deposit; osteoporosis is not uncommon. The clinical diagnosis may be difficult during pregnancy because characteristically
similar striae are seen in both pregnancy and Cushing's
syndrome. Comparisons of close-up photographs from earlier years are
helpful in detecting subtle changes. The diagnosis is confirmed by
the proper use and interpretation of tests assessing the hypothalamic - pituitary - adrenal
axis. A significant and persistent elevation in
urinary free cortisol is the best indication of cortisol overproduction. It
should be kept in mind, however, that an elevation does occur in
normal pregnancy. Mean urinary free cortisol levels of 127 μg/d (range, 68 – 252 μg/d), as compared with nonpregnant levels of 37 μg/d (range, 11 – 83 μg/d), have been reported.140 Plasma cortisol increases threefold from the first to the third trimester
of pregnancy. Random determinations of plasma cortisol are of little
help, because values are normal in many patients with Cushing's
syndrome. Furthermore, in pregnancy it is normally elevated because
of an increase in serum CBG. The overnight dexamethasone suppression test, although
an excellent screening test in nonpregnant women, is of
limited value in pregnancy. Lack of diurnal variation for cortisol is
suggestive of the disease, but to properly assess the results of the test, specimens
should be obtained between 7 AM and 9 AM and late in the
evening. A random plasma cortisol 4 PM value could be misleading. Hence, in
pregnancy the determination of free cortisol in a 24-hour urine
specimen is the best screening test to rule out Cushing's syndrome. Concomitant
determination of creatinine in the urine is necessary
to assure proper urinary collection. Once the diagnosis of Cushing's
syndrome is suggested, the cause should be investigated. As mentioned
earlier, bilateral adrenal hyperplasia and adrenal adenomas are the
most common causes in pregnancy; adrenal carcinoma is rare. Plasma
ACTH levels are helpful when interpreted in the light of urinary cortisol
values. High-normal or elevated values in the presence of high urinary
cortisol is suggestive of ACTH-dependent Cushing's syndrome (Cushing's
disease). Low ACTH values are consistent with unilateral
adrenal tumor. Further delineation of the pathologic cause is accomplished by use of the
dexamethasone suppression test. For the test, urinary cortisol is measured
before and during 2 days each, first with a small dose (0.5 mg
every 6 hours) and then with a high dose of oral dexamethasone (2 mg
every 6 hours). Patients with Cushing's syndrome, regardless of the
cause, fail to suppress urinary free cortisol with the small doses (0.5 mg
every 6 hours). Patients with bilateral adrenal hyperplasia show
a 50% or greater fall in urinary free cortisol with dexamethasone given
in the high-dose regimen, whereas there is no suppression in patients
with adrenal tumor. Exceptions to this type of response are occasionally
encountered.141 These tests have been useful in pregnancy. In patients with Cushing's disease, an MRI of the pituitary gland
may identify a tumor. However, one must keep in mind the enlargement of
the pituitary gland that occurs during normal pregnancy. Selective petrosal
sinus sampling, a procedure used in localization of the pituitary
lesion, is not indicated in pregnancy because of radiation exposure. For
cases in which the laboratory tests are suggestive of adrenal lesion, an
MRI may delineate the adrenal pathology. Cushing's syndrome itself is an uncommon disorder and is seen rarely
during pregnancy. No more than 100 cases have been reported.142 Spontaneous exacerbations143,144,145 and amelioration139,146,147 of symptoms during pregnancy or postpartum have been reported. Wallace
and colleagues142 reported on a patient with recurrent Cushing's syndrome in three
successive pregnancies. In this woman with “pregnancy-induced Cushing's
syndrome,” hormonal and clinical features normalized
a few weeks postpartum. Serum ACTH was suppressed early in pregnancy, and
there was a lack of urinary cortisol suppression with the administration
of dexamethasone. Although the authors were unable to detect
any direct adrenal stimulation in vitro and in vivo from extracts of placenta
or maternal serum, the clinical course is suggestive of a stimulator
originating in the fetal placental unit. The clinical course resembles
reports of periodic Cushing's syndrome. Maternal and perinatal morbidity and mortality are significant in Cushing's
syndrome. A few maternal deaths in the postpartum period caused
by congestive heart failure have been reported,148 and one death was caused by disseminated aspergillosis in a mother with
ectopic ACTH syndrome.138 Arterial hypertension occurs in over 69% of cases,136 overt diabetes in 30%, preeclampsia in 7%, congestive heart failure in 7%, and
maternal death in 4%. Prematurity has been reported in 52% of
infants, and fetal wastage is close to 30% (abortions, stillbirth, and
neonatal death).142 Intrauterine growth retardation has been reported in 25% of patients with
Cushing's syndrome.137,149 Medical treatment of Cushing's syndrome during pregnancy has been
reported; in several cases metyrapone was used as the sole form of treatment.142,149,150,151 Metyrapone acts primarily to inhibit steroid 11β-hydroxylation, therefore
decreasing the secretion of cortisol by the adrenal glands. Its
main use is as a diagnostic tool for assessing ACTH reserve, and it
is also used in the diagnosis of Cushing's syndrome. It has been
used as a therapeutic agent in nonpregnant patients with Cushing's
syndrome for a short period of time before surgery or after radiation
therapy to the pituitary gland. In the case reported by Gormley and
co-workers,149 fetal growth and fetal well-being were satisfactory and the newborn showed
no signs of adrenal insufficiency. Metyrapone in doses of 250 to 1000 mg
daily was given from 29 weeks' gestation until spontaneous onset
of labor at 36 weeks' gestation. Hypertension was controlled with α-methyldopa 500 mg
every 6 hours. The authors reported clinical improvement
with a reduction in back pain, peripheral edema, and facial
plethora. In another case caused by adrenal carcinoma,150 the patient developed preeclampsia a few days after metyrapone therapy. The
infant was born prematurely and died a few days later. In the case
reported by Wallace and colleagues,142 metyrapone was given in the second and third pregnancies; the second terminated
at 32 weeks' gestation because of placenta abruptio, and the
third terminated at 37 weeks' gestation. In the third pregnancy, dexamethasone
was added at 22 weeks of pregnancy to avoid adrenal insufficiency
caused by the use of metyrapone. The infant was born with adrenal
suppression, coarctation of the aorta, and nephrocalcinosis. Two cases
were treated with cyproheptadine, a serotonin antagonist, with delivery
of normal infants.152 One case of adrenal carcinoma was treated with ketoconazole.153 In 11 cases, adrenal surgery was performed during pregnancy, most during
the first half of pregnancy and a few during the last half.148 Patients who had surgery performed during the last half of pregnancy (bilateral
adrenalectomy for hyperplasia or unilateral adrenalectomy for
adenoma) had a significantly better pregnancy outcome, with fewer fetal
losses (1 of 11 versus 9 of 30 pregnancies) and less prematurity. Pricolo
and associates154 reported on transsphenoidal pituitary surgery performed in a woman at 22 weeks' gestation. The
pregnancy had to be terminated at 30 weeks' gestation
because of severe hypertension.155 In a recent report, transsphenoidal surgery was performed in the second
trimester of pregnancy, and a healthy infant was delivered at 38 weeks' gestation.156 A few cases of Nelson's syndrome (pituitary tumor with skin pigmentation
after bilateral adrenalectomy for Cushing's disease) have
been reported during pregnancy with good infant outcome.157,158 Because ACTH does not cross the placenta, the fetal adrenal gland is not
affected. In four cases, melanosis appeared as the first sign of pituitary
tumor during pregnancy, suggesting that pregnancy is an additional
predisposing factor in the development of Nelson's syndrome.157 These patients, as in patients with prolactinomas, should be followed
carefully during pregnancy with visual field examinations to detect any
enlargement of the pituitary gland. Addison's Disease Addison's disease, or primary adrenal insufficiency, results from
total destruction of the adrenal cortex. In cases of adrenal insufficiency
secondary to ACTH deficiency, the zona glomerulosa is preserved; therefore, there
is normal secretion of aldosterone. The most common cause
of primary adrenal insufficiency is autoimmune disease of the adrenal
glands, which occurs in about 70% of cases of Addison's disease. The
second most common cause is tuberculosis. Other unusual causes
include surgical bilateral adrenalectomy for systemic diseases, bilateral
metastatic carcinoma, bilateral adrenal bleeding during anticoagulant
therapy, and, rarely, fundal infections. Severe systemic infections
by meningococcus (Waterhouse-Friderichsen syndrome) may present as
acute adrenal insufficiency characterized by vascular collapse and petechial
hemorrhage in the skin and mucosa. With the wide use of corticosteroids for the treatment of systemic diseases, adrenal
atrophy due to ACTH suppression is perhaps the most common
cause of adrenal insufficiency. In these cases, adrenal insufficiency
is caused by chronic suppression of pituitary ACTH. Patients receiving
corticosteroid therapy for various periods of time may develop relative
adrenal insufficiency. This may manifest for the first time during
periods of stress, at which time, because of chronic ACTH suppression, the
adrenal glands are unable to respond with an increase in cortisol
secretion. It has been estimated that it takes an average of 8 months
for the hypothalamic - ACTH - adrenal axis to recover completely after
the discontinuation of corticosteroid therapy.159 The most common symptoms in patients with chronic primary adrenal insufficiency
are weakness, fatigue, and weight loss. Characteristically, these
patients feel relatively well in the morning and experience increased
fatigue and asthenia as the day progresses. Blood pressure is low, and
patients may complain of postural hypotension. Gastrointestinal
symptoms, such as nausea, anorexia, and sometimes vomiting and diarrhea, are
not uncommon and may result in weight loss. Although pigmentation
of the skin and mucosa is characteristically described in patients
with Addison's disease, it is not seen in all such patients. The
pigmentation is usually seen in body creases, such as palms of the hands, knuckles, knees, and
elbows; in scars; and in the genital, gingival, and
buccal areas. Vitiligo may be found in 10% to 15% of patients with
Addison's disease. These lesions have been ascribed to autoimmune
destruction of the melanocyte and occur in hyperpigmented areas. Gonadal
function is disturbed, and in women, axillary and pubic hair may
be reduced because of lack of adrenal androgens. Adrenal crisis is
characterized by severe hypotension, gastrointestinal symptoms such as
nausea, vomiting, and diarrhea, and severe dehydration. These patients
may present with elevated body temperature, although infection is not
always demonstrated. Typical laboratory findings in patients with Addison's disease include
mild anemia, hyponatremia, hyperkalemia, and an increase in serum
blood urea nitrogen. Serum electrolytes may remain within normal limits
in some cases. Hypercalcemia may be seen during the acute phase of
adrenal insufficiency. Hypoglycemia may be found in patients in the fasting
state and during adrenal crisis. Chest radiographs may demonstrate a small heart, and the electrocardiogram
may show changes related to electrolyte disturbance. The diagnosis is confirmed by a lack of cortisol response to continuous
ACTH administration or elevated serum ACTH levels in the presence of
low serum cortisol concentrations, or both.160 ACTH STIMULATION TEST. Baseline or random serum cortisol levels are low in most patients with
Addison's disease. However, in some patients the adrenal glands are
still able to respond to the high levels of circulating plasma ACTH, and
these patients may have normal cortisol levels on a random specimen. On
the other hand, low plasma cortisol levels are not diagnostic
of adrenal insufficiency, because they are seen in many other situations
in which the adrenal glands are not diseased. Furthermore, in pregnancy
or in patients on estrogen therapy, plasma cortisol is elevated because
of an increase in plasma CBG concentrations. Therefore, the diagnosis
of adrenal insufficiency is confirmed by dynamic tests, namely, serum
cortisol response to the administration of ACTH. Short ACTH Stimulation Test. This is the simplest test available to rule out the presence of Addison's
disease. For the test, synthetic ACTH (cosyntropin) 0.25 mg is
given as a rapid intravenous bolus, and plasma cortisol is measured before
administration and 30 and 60 minutes after. A peak cortisol response
of 18 μg/dL over the baseline values rules out Addison's
disease. However, a lack of response does not confirm the diagnosis, and
a more prolonged ACTH stimulation test is needed.161 Long ACTH Stimulation Test. There are several modifications of the original test. The basis for the
test is the continuous administration of ACTH for several days along
with measurement of plasma or urinary corticosteroids.162 For the test, ACTH in the form of cosyntropin, 0.25 mg in 500 mL 5% dextrose
in normal saline, is given every 12 hours for 48 hours, and serum
cortisol is measured before administration and every 12 hours thereafter. An
increase in serum plasma cortisol of over 30 μg/dL rules
out the presence of primary adrenal insufficiency. To prevent an acute
adrenal crisis in patients with significant symptoms of adrenal insufficiency, dexamethasone, 0.5 mg, can be added to the continuous ACTH infusion. This
small amount of corticosteroid does not interfere with the
determination of serum cortisol and provides enough glucocorticoid
to correct the patient's symptoms. This 48-hour continuous ACTH infusion
can accurately distinguish between primary and secondary adrenal
insufficiency. PLASMA ACTH DETERMINATION. Patients with cortisol deficiency caused by primary adrenal disorders have
elevated concentrations of ACTH; these can be measured through a radioimmunoassay
technique.163,164 Normal values early in the morning are between 20 and 150 pg/mL. Baseline
cortisol values less than 10 μg/dL and ACTH levels of greater than 250 pg/mL
are diagnostic of primary adrenal insufficiency. Treatment Patients with primary adrenal insufficiency require treatment for the remainder
of their lives, with occasional exceptions. Cortisone or hydrocortisone
is the treatment of choice, in doses of 20 to 30 mg/day, with
two thirds of the dose being given in the morning and one third in
the evening. In most patients a mineralocorticoid is added in the form
of fluorohydrocortisone. The daily amount has to be adjusted for each
patient, and the total dose varies from 0.05 to 0.2 mg. Although cortisol
is the natural hormone produced by the adrenal gland, and it is the
drug of choice, other synthetic glucocorticoids may be used. Prednisone
and prednisolone are less expensive and more available than cortisone, and
either one can be used in doses of 3 to 5 mg in the morning
and 1 to 3 mg in the evening. In cases of moderate stress, such as fever
or gastrointestinal disturbances, the dose of corticosteroids should
be doubled for a few days. Adrenal crisis is an emergency requiring immediate treatment. Intravenous
fluids in the form of normal saline and glucose should be given, and 100 mg
hydrocortisone should be injected as a bolus intravenously. An
additional 200 mg should be given during the first 24 hours. A search
for the cause of the acute crisis should be made, because in most cases
infection is responsible. Patients with known adrenal insufficiency and patients on corticosteroid
therapy undergoing selective surgery should be given cortisol in total
doses of 200 to 300 mg during the day of surgery. Many regimens have
been proposed for the management of these patients, but the one that
I recommend is the use of cortisol, 100 mg intravenously every 8 hours
in continuous infusion for the first 24 hours and then decreased by 50 mg/d
until the patient is able to take oral medications. The infusion
should be started early in the morning on the day of surgery. In most
cases the patient is able to return to a routine daily dosage by the
fourth or fifth day postsurgery. Because the amount of cortisol given
has enough mineralocorticoid action, no mineralocorticoid supplementation
is needed during this time. Daily determinations of serum electrolytes
and glucose should be obtained, because this amount of cortisol
may produce hypokalemia and hyperglycemia. To supply enough corticosteroids, it
is suggested that an injection of cortisone acetate, 50 to 100 mg
intramuscularly, be given the day before surgery. The same regimen
is applied to patients who received corticosteroid therapy in the
past and discontinued it within 1 year. An additional regimen involves
cortisone acetate, 50 mg intramuscularly, every 6 hours the first day, every 8 hours
the second day, and every 12 hours the fourth day after
a surgical procedure. During surgery, 100 mg of hydrocortisone is added
to the intravenous drip. Pregnancy in patients with known Addison's disease has resulted in
normal infants. Early reports have indicated no increase in miscarriage
rate, prematurity, or intrauterine death.165,166 In general, the amount of glucocorticoid needed is the same as in nonpregnant
patients, with the requirement increasing during labor. Patients
with Addison's disease should be managed during labor in the same
way as patients under stressful situations. Fasting serum glucose
is lower during pregnancy, and patients with untreated adrenal insufficiency
have a tendency to develop hypoglycemia. Severe hypoglycemia has
been reported in a few patients with ACTH deficiency during pregnancy9 and may occur in patients with Addison's disease if not enough corticosteroids
are given or if the patient omits a dose. Patients with
chronic adrenal insufficiency should be instructed to take the medications, particularly
in cases of nausea and vomiting. They should have injectable
corticosteroids at home for such cases. Dexamethasone phosphate
in 4-mg injectable syringes is available. Patients should always carry
with them identification stating their diagnosis and medication. It
has been reported that patients may need an increased amount of mineralocorticoids
during pregnancy because of the high levels of progesterone, which
have a sodium-losing effect. Elevated levels of serum aldosterone
are characteristic in normal pregnancy and have been attributed
to the high levels of progesterone that are also present. Infants of mothers who receive corticosteroid therapy rarely develop adrenal
insufficiency. In a review of 260 pregnancies in which the mother
took steroids for variable periods, only one infant was believed to
have adrenal insufficiency.167 Mothers on chronic corticosteroid therapy have low urinary or plasma estriol
levels. As mentioned before, occasionally an infant is reported
to be born with adrenal insufficiency as a result of maternal steroid
therapy.168 Congenital Adrenal Hyperplasia Congenital adrenal hyperplasia (CAH)169 is a hereditary disorder involving both adrenal glands. It is produced
by a number of defects of corticosteroid biosynthesis that result in
a reduced production of cortisol and a secondary increase in ACTH secretion, which
is then responsible for adrenal hyperplasia and an accumulation
of hormonal precursors. Defects of several enzymes, such as cytochrome
P-450 oxidase, may produce different clinical pictures (Fig. 2). 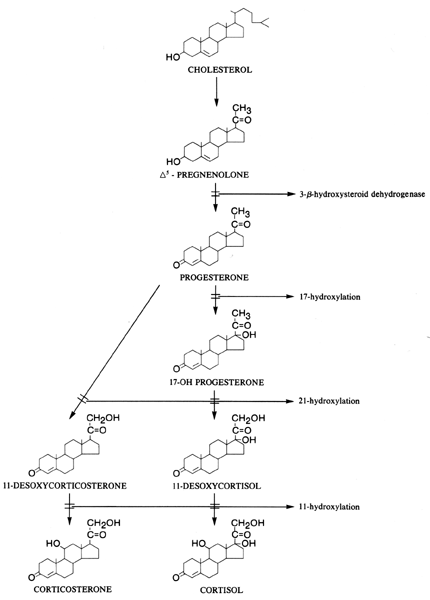 Fig. 2. Biosynthetic defects in corticosteroid synthesis. Fig. 2. Biosynthetic defects in corticosteroid synthesis.
|
The most common defect is the 21-hydroxylation defect, which is found in
more than 90% of patients with congenital adrenal hyperplasia. It is
an autosomal recessive disorder involving a gene on the short arm of
chromosome 6. It affects about 1 in every 14,000 live births, but in some
populations it is more common (1 in 80 births).170 The clinical manifestations depend on the severity of the abnormality
in the 21-hydroxylase genes. Women with severe deficiency of the enzyme (the
classical form of 21-hydroxylase deficiency) have a low fertility
rate because of oligoanovulation. Abnormal genitalia and sodium wasting
are recognized at the time of delivery, as are signs of virilization. In
the milder form of the disease, virilization is the only manifestation, because
aldosterone secretion is not seriously affected. The
third form of the disease is mild and is detected in childhood or adulthood.171 Other types of defects include the 11-hydroxylation defect and, rarely, the
defect of the 3-β-hydroxysteroid-dehydrogenase enzyme. The goals of treatment are to prevent adrenal insufficiency, to provide
salt-retaining corticosteroids in the salt-losing types of cases, and
to suppress androgen secretion to prevent virilization and abnormalities
of growth. This is achieved with doses of hydrocortisone of 25 to 75 mg/d
or 25 mg/m2/d. Dexamethasone with a longer half-life is preferred by some in doses
of 0.25 to 0.75 mg/d.172 Parameters of adequate glucocorticoid dosage include determination of
plasma 17-hydroxyprogesterone (17-OHP), androstenedione, and testosterone. Patients
with the salt-losing variety also require mineralocorticoid
therapy in the form of Florinef (fludrocortisone) 0.05 to 0.3 mg/d; the
dose is adjusted to normalize plasma renin activity. No adjustment
is needed in pregnancy.173 The hormonal markers for therapy monitoring change during pregnancy, with
the exception of the determination of maternal serum free testosterone, which
is useful for the assessment of adequate therapy.173 Klingensmith and co-workers174 reported a group of 18 females, 10 years of age or older, with congenital
adrenal hyperplasia followed for many years. Initiation of menstruation
was delayed in all of these patients when compared with the general
population. Eight patients were treated for the first time after age 20 years, and
none of them were able to conceive. Of 20 patients in
whom treatment was started between ages 6 and 20 years, 64% who were
sexually active had at least one pregnancy. In patients first treated
before age 6 years, the pregnancy rate was about 60%. The amount of cortisone
needed was between 30 and 56 mg/m2 in 24 hours, and the levels of urinary 17-ketosteroids were between 2.5 and 5.3 mg/d. All
women were maintained during pregnancy on the prepregnancy
dose of steroids. Of the 13 pregnancies in these patients, 9 were
viable and only 1 resulted in an infant born with multiple congenital
malformations, but the mother had systemic lupus erythematosus and
was treated with large doses of salicylate. All of the mothers underwent
cesarean section, and there was only one case of a premature infant
with mild residual cerebral palsy. No patient in this study had an
infant with CAH. However, the fertility rate was low and first-trimester
abortions were high.173,174,175,176 Prenatal evaluation of the partner of a woman who has the disease or is
a carrier is important in evaluating for possible fetal involvement. The
partner has a 1% to 6% chance of being a carrier of the disease. Determination
of plasma 17-OHP at baseline and after the administration
of ACTH allows, in most cases, detection of the partner's heterozygous
state.169 Fetuses are considered “potentially affected” if: (1) both
parents are heterozygous, (2) both parents are homozygous, or (3) one
parent is homozygous and the other heterozygous. Prenatal diagnosis of CAH due to 21-hydroxylase deficiency is possible
with the detection of elevated amniotic fluid 17-OHP and adrenal androgen
concentrations or with linkage analysis and human leukocyte antigen
typing of cultured amniotic fluid cells. However, because these tests
cannot be completed before 16 to 17 weeks' gestation, any preventive
therapy should be given before the results of the tests become available, because
masculinization of the fetus has occurred between 10 and 16 weeks' gestation.177,178 Chorionic villus biopsy at 8 to 10 weeks' gestation allows for the most
accurate prenatal diagnosis using molecular genetic techniques.179,180 However, the fetal loss rate after chorionic villus sampling (CVS) is 1.3% to 1.6%.173 Early treatment, before 6 weeks' gestation, may prevent the development
of masculinization of external genitalia in the female-affected fetus. Dexamethasone
is the drug of choice because it readily crosses the
placenta (compared with all the other glucocorticoids) and suppresses
fetal adrenal androgen synthesis, reducing masculinization. Mothers who
are potentially affected should be treated until fetal sex and involvement
are known. The treatment is effective in about two thirds of patients.181,182 The dose of dexamethasone is 1 to 1.5 mg/d from early in pregnancy until
delivery (20 μg/kg maternal weight). Maternal estriol levels are
an accurate index of fetal adrenal function and genital outcome.183 Pang and colleagues184 reported maternal side effects of this therapy in three women. Cushingoid
facial features, facial hirsutism, excessive weight gain, permanent
scarring of striae, hypertension, and gestational diabetes were the
most common complications of therapy. The authors recommended that future
patients should be informed about potential side effects, mothers
receiving treatment should be closely monitored, medical intervention
should be provided when indicated, and the dose of dexamethasone should
be reduced during the second half of pregnancy, to reduce side effects. In the last 10 years, direct molecular analysis of the 21-hydroxylase (CYP21) gene
has been performed since CVS became available in 1984. CVS
can be performed at 10 weeks' gestation. Mercado and associates185 published their evaluation of 258 cases from 235 families with a sibling, parent, or
close relative with 21-hydroxylase deficiency. They followed 229 pregnancies
that resulted in live births. Prenatal diagnosis
was made by amniocentesis; 17-OHP was measured in amniotic fluid, or
human leukocyte antigen genetic linkage marker analysis was used. The
accuracy rate of each diagnostic procedure was 95%. In their protocol, treatment
was started with dexamethasone 20 μg/kg/d in two or three
divided doses before 10 weeks' gestation in those mothers whose babies
could potentially be affected. Treatment was discontinued if the fetus
was found to be male or an unaffected female. Prenatal treatment was
initiated in 101 mothers, and in 22 of them the fetus was confirmed
to be affected with classical 21-hydroxylase deficiency. There were 13 female
and 9 male infants. Nine females affected were treated throughout
pregnancy; four were normal at birth and five were virilized but
to a lesser degree than their siblings. Of 75 cases in which the mothers
refused treatment, 15 were affected, 8 of them females. Of these 8 females, there
were three elective abortions, and all five that went to
term had virilized genitalia. The authors concluded that dexamethasone
administered at or before 10 weeks' gestation was effective in reducing, and
in some cases preventing, virilization. Follow-up of the 84 newborns
prenatally treated showed normal birth weight, birth length, and
head circumference compared with untreated fetuses. There is an ongoing
evaluation of the long-term effects of dexamethasone-exposed fetuses.185 11-Hydroxylase deficiency is common in the Middle East but rare in European
populations.186 The production of cortisol is reduced, and there is an excessive production
of 11-deoxycortisol, 11-deoxycorticosterone, and adrenal androgens.169 Hypertension, hypokalemia, and virilization are the most common form of
presentation, along with infertility. The diagnosis may be made for
the first time in adolescence or adulthood, when the patient presents
with infertility, or oligomenorrhea, hirsutism, and short stature. Treatment
with glucocorticoids normalizes infertility, and successful pregnancies
have been reported.187 Primary Aldosteronism Primary aldosteronism was described by Conn in 1955188 and is characterized by hypertension and hypokalemia due to excessive
production of aldosterone by the adrenal glands. The first cases described
were of patients with single adrenal adenomas, but now it is known
that about 20% to 30% of patients with primary hyperaldosteronism suffer
from bilateral adrenal hyperplasia or multiple microadenomas; rare
cases of corticosteroid suppressive hyperaldosteronism, adrenal carcinoma, and
congenital primary aldosteronism have been reported. Primary hyperaldosteronism is a rare disorder accounting for less than 0.5% of
all causes of hypertension. Symptoms such as headaches and muscular
weakness are present in some patients, the latter being a complication
of hypokalemia. Serum potassium is decreased in more than 90% of
the cases. Metabolic alkalosis with hypochloremia and an elevation in
serum bicarbonate is commonly seen. Urinary potassium excretion is elevated
and inappropriate to the low serum potassium levels. Renal function
is within normal limits, and the pH of the urine is neutral or alkaline. Patients
with marked hypokalemia may complain of polyuria due
to a renal tubular defect characterized by being Pitressin resistant. The diagnosis is suspected in those patients with hypertension and hypokalemia; all
other causes of hypokalemia, particularly ingestion of diuretics, must
be ruled out. High levels of urinary potassium in the presence
of hypokalemia are very suggestive of primary hyperaldosteronism. The
diagnosis is confirmed by (1) an elevation in serum or urine aldosterone, (2) suppressed
levels of plasma renin, (3) a lack of plasma
renin elevation on administration of a potent diuretic such as furosemide, and (4) failure
of the aldosterone level to be suppressed by the
administration of 200 μg 9-α-fludrocortisone twice a day for 3 days.189 In normal pregnancy there is an increase in aldosterone secretion and plasma
aldosterone levels starting in the first trimester; this peaks at 36 weeks' gestation
at a level eightfold to tenfold higher than that
in nonpregnant women. This elevation in plasma aldosterone is normally
suppressed in pregnancy by the administration of 9-α-fludrocortisone.190 Plasma renin levels increase early in pregnancy, reaching a peak at 12 weeks
and remaining elevated throughout pregnancy. In the few cases of primary hyperaldosteronism reported in pregnancy, the
aldosterone levels were elevated and the plasma renin was suppressed.190,191,192,193 In the case reported by Gordon and Tunny,191 surgery was performed during pregnancy with good results. In the case
reported by Biglieri and Slaton,192 the metabolic and blood pressure abnormalities improved spontaneously
during pregnancy. A case of virilization in the fetus has been reported.193 In the reported cases of primary hyperaldosteronism in pregnancy mentioned
above, metabolic alkalosis was present instead of respiratory alkalosis, which
is characteristically seen in normal gestation. Serum bicarbonate
in plasma is normal or elevated, in contrast to the low value
seen in normal pregnant women. Treatment is based on the cause of aldosteronism. An MRI of the abdomen
is effective in localizing the adrenal lesion. In most cases reported
in pregnancy, a single adrenal tumor is the cause, in which case adrenalectomy
is the treatment of choice. In cases of bilateral adrenal hyperplasia, medical
treatment with spironolactone is effective.186,194 Pheochromocytoma Pheochromocytoma is a rare syndrome accounting for about 0.5% of all cases
of newly diagnosed hypertension. It occurs with equal frequency in
both sexes and is more common in the third and fourth decades of life. In
Northern Ireland, the annual incidence of pheochromocytoma is 1 case
per 750,000 population, half of them women. In a 21-year period, seven
cases occurred during pregnancy.195 In most cases the disease is produced by a single adrenal adenoma; about 15% are
multiple, and 10% to 12% are malignant. Involvement of both
adrenal glands is seen in cases with a strong family history of pheochromocytoma. Furthermore, it may be part of the syndrome of multiple endocrine
neoplasia type II (MEN type IIA and IIB) in association with
medullary carcinoma of the thyroid and parathyroid adenoma (Sipple's
syndrome). Pheochromocytoma may also be associated with neurofibromatosis, and
it is estimated that 5% of patients with neurofibromatosis
will develop a pheochromocytoma. In fewer than 10% of cases, the chromaffin
tumors initiate from outside the adrenal gland in the organ of
Zuckerkandl196 and the bladder wall197 and are named paragangliomas. Hypertension is the most common symptom; it can be persistent or paroxysmal. In
addition to hypertension, headaches, sweating, palpitations, nervousness, tremor, weakness, abdominal pain, and warm flashes are not
uncommon. Some patients have markedly labile blood pressure. The attacks
can be precipitated by induction of anesthesia, examination of the
abdomen, or changes in position. The patient with pheochromocytoma may
be hypermetabolic, with weight loss, nervousness, and elevation of
the basal metabolic rate. Glycosuria and mild intolerance to glucose are
not unusual. Pheochromocytoma associated with pregnancy is life-threatening for mother
and fetus. In 1969, Fox and co-workers198 described 1 case and reviewed 89 cases reported in the literature. In 1982, Schenker
and Granat199 reviewed 112 pregnancies occurring in 89 patients. They emphasized the
poor prognosis for mother and fetus. The maternal mortality rate was 48% and
the fetal mortality rate was 54% when the disease remained undiagnosed
during pregnancy; high mortality occurred mainly within 72 hours
of delivery. The cause of death included cerebrovascular accidents, cardiac
arrhythmias, shock, and acute pulmonary edema. Only 1 of 26 patients
died when the diagnosis was made before delivery. This stresses
the importance of early diagnosis and effective therapy. In 17 cases
the symptoms disappeared between pregnancies, and in two thirds of the
cases pheochromocytoma occurred in multiparous women. Hypertension (paroxysmal or sustained) was present in 82% of the patients, headaches
in 66%, palpitations in 17%, anxiety in 15%, and convulsions
in 10%. In several cases, sudden shock appeared spontaneously or
was induced by anesthesia or vaginal delivery in patients without previous
symptoms (Table 7). In 22 patients, the diagnosis was made for the first time during pregnancy. Of 67 patients
in whom pheochromocytoma remained undiagnosed during
pregnancy, acute toxemia was diagnosed in 29 (43%) and essential
hypertension was diagnosed in 11 (16.4%). TABLE 7. Symptoms of Pheochromocytoma in Pregnancy
| Cases(%) |
Hypertension | 90 |
Headaches | 70 |
Palpitations | 40 |
Sweating | 35 |
Anxiety | 30 |
Blurred vision | 20 |
Convulsions | 10 |
Dyspnea | 10 | (Adapted from Schenker JG, Granat M: Phaechromocytoma and pregnancy - An
updated appraisal. Aust NZ J Obstet Gynaecol 22:1, 1982)Fetal wasting is high in patients with pheochromocytoma. Of 162 pregnancies, the
fetal loss was 53%; spontaneous abortion occurred in 12%, intrauterine
death in 23%, and neonatal or intrapartum death in 18%. Fetal
mortality has improved since the introduction of α-adrenergic receptor
blocking agents.200 The diagnosis of pheochromocytoma is confirmed by the demonstration of
elevated levels of catecholamines or their metabolites (norepinephrine
and epinephrine), methylated metabolites (normetanephrine and metanephrine), and
the methylated deaminated metabolite vanillylmandelic acid (VMA). An
elevation in VMA, catecholamines, or metanephrine in a 24-hour
urine specimen is suggestive of the diagnosis. This should be confirmed, however, by
a second 24-hour collection. The following recommendations
have been suggested for the proper interpretation of urinary catecholamine
and its metabolites: - Twenty-four-hour urine collections are preferable to random ones; urinary
creatinine should be measured in each 24-hour collection to assess
its adequacy.
- It is preferable to collect the urine when the patient is at rest, is on
no medication, and has not had recent exposure to radiographic contrast
media. Among the medications for hypertension, diuretics, adrenergic
blocking agents, and hydralazine do not interfere appreciably.
- Urine collection must be acidified (pH below 3.0) and kept cold during
and after collection.
- The diagnostic information is greater in a patient with paroxysmal symptoms
if a 24-hour collection is done when he or she experiences the crisis.
Total urinary free catecholamines are elevated in most patients with pheochromocytoma. Normal
total excretion is between 100 and 150 μg/d, which
is similar to that of the nonpregnant state. However, it may be
slightly elevated in preeclampsia201 and also in stressed, working normal pregnant women.202 Specific quantitation of epinephrine and norepinephrine may assist in
the localization of a lesion; increased epinephrine excretion in excess
of 50 μg/d suggests an adrenal lesion. Excessive secretion of noradrenaline
is suspicious, but not diagnostic, of extra-adrenal tumor. False-positive
elevations may occur in patients who are taking methyldopa, are
undergoing strenuous exercise, have increased intracranial pressure, have
hypoglycemia, are undergoing clonidine withdrawal, or are
undergoing aminophylline administration. Plasma levels of catecholamines
can be measured, particularly during a paroxysmal episode. It is important
to adhere strictly to the technique of blood collection. Value
fluctuations are common under routine stressful situations, such as
exercise, venipuncture, and anxiety. It is recommended that the patient
rest in the supine position in a quiet environment for 30 minutes before
blood is drawn. Blood is obtained through an indwelling catheter. The
plasma should be centrifuged and the serum stored at temperatures
lower than 4°C until assayed.203 As soon as the diagnosis is confirmed, administration of α-adrenergic
blocking agents should be started.204,205 The drug most often used is phenoxybenzamine. Starting with 10 mg/d, the
dose can be increased by 10 mg every 2 to 3 days until blood pressure
and symptoms are controlled. The blocking dose is 1 mg/kg/d. It may
need to be adjusted because it has a half-life of about 24 hours and
hence is cumulative. The usual dose of phenoxybenzamine is 40 to 80 mg/d.206 Prazosin in doses of 6 to 10 mg has been used as a selective α1 antagonist in pregnancy.207 In addition, α-adrenergic receptor blockade increases the blood volume
and may improve congestive heart failure as well as angina pectoris, if
present. A 2-week course of an α-adrenergic receptor blockade
is the cornerstone of preoperative management,208 although a 3-day course has been used successfully in pregnancy.209 Beta-blockers are added to the regimen when tachycardia develops after
the administration of an α-adrenergic blocking agent.204,210 Propranolol 10 mg three to four times a day is useful in maintaining the
pulse rate between 80 and 100 beats a minute. Hypertensive crises are
treated with intravenous phentolamine in doses of 1 to 5 mg. Intravenous
therapy can also be used in cases of unstable hypertension in preparation
for surgery.211 Alpha blockage should be completed before surgery is contemplated; it
takes 10 to 14 days to achieve an acceptable result. Indicators of adequacy
include a blood pressure less than 165/90 mmHg; the presence of
orthostatic hypotension, but with standing blood pressure greater than 90/45 mmHg; no
changes in the electrocardiogram for the last 2 weeks (no
ST-segment or T-wave abnormalities); and less than one premature ventricular
contraction every 5 minutes.212 Surgical intervention is the treatment of choice in cases of pheochromocytoma. Tumor
localization in 47 pregnant patients showed that 25% were
in the right adrenal gland, 17% were in the left, 17% were bilateral, and 32% were
intra-abdominal extra-adrenal.201,213 Localization of the tumor is imperative and is achieved in most cases
with modern radiologic techniques. In pregnancy, CAT scans, arteriography, and
selected venous catheterization are contraindicated for obvious
reasons. MRI is useful in localizing and diagnosing adrenal lesions. In
pregnancy, this is the method of choice for localization of the tumor. The
major advantages are lack of ionizing radiation, lack of contrast
dye, and the ability to distinguish pheochromocytomas from other
adrenal lesions by the high-intensity signal emitted in T2-weighted images. Ultrasonography is of little value; pressure exerted
by the sonogram probe has been reported as the causative agent in a paroxysmal
attack.214,215,216 When the diagnosis is made during the first trimester, elective termination
of pregnancy followed by tumor excision is recommended by some,206 whereas selective tumor resection is recommended by others.199,209,217 However, the incidence of spontaneous abortion occurring after surgery
is high. During the last two trimesters of pregnancy, medical treatment is the treatment
of choice, with removal of the tumor at the time of cesarean
section.215,216 Burgess218 reviewed 42 cases from the literature in which the diagnosis was made
antepartum; the maternal mortality rate was 12% and the fetal mortality
rate was 40%. The best results are obtained when the pregnancy has advanced
to viability, the fetus is delivered by cesarean section, and
tumor exploration is done immediately after. Cesarean section rather than
vaginal delivery is strongly recommended as the safest procedure for
both mother and fetus.199 In Burgess' study, the fetal mortality rate was reduced to 17% when the
diagnosis was made in the first two trimesters of pregnancy. For patients
in whom the diagnosis was made in the third trimester, the fetal
mortality rate was 0%, compared with 42% in the group that did not undergo
administration of an α-adrenergic blocking agent. No maternal
mortality has been reported in patients treated with an α-adrenergic
blocking agent. Stenstrom and Swolin209 reviewed 29 cases from the literature and 3 of their own in which the
pheochromocytoma was diagnosed antepartum and treated with α-adrenergic
receptor blockade. Fourteen patients underwent short treatment of 4 to 21 days, and 6 underwent treatment of 46 to 91 days; in 9 cases
the duration was unknown. The investigators recommended the use of elective
tumor resection in the first trimester and medical treatment followed
by cesarean section and tumor removal when the diagnosis is made
after the first trimester. The fetal mortality rate in the treated cases
was 19% compared with 50% in those without the benefit of α-adrenergic
receptor blockade treatment. An anesthesiologist should be consulted
and become closely involved in the management of patients with
pheochromocytoma.211,219 Virilization Virilization (Table 8) in pregnancy is exceedingly rare. Several cases have been described with
normal fetal outcome. The most common cause is luteoma of pregnancy, which
is defined as a benign human chorionic gonadotropin (hCG) - dependent
ovarian tumor that develops during pregnancy. Virilization occurs
in about 25% of such patients. High levels of testosterone and androstenedione
are responsible for virilization. These patients develop
hirsutism, acne, deepening of the voice, and clitoral enlargement, beginning
early in the second trimester. Fetal virilization is seen in 65% of
females born to patients whose virilization began during pregnancy. Regression
with normalization of the elevated androgen levels occurs
in the postpartum period. In the case reported by Thomas and co-workers,220 virilization had occurred in previous pregnancies; chorionic gonadotropin
stimulation 8 months postpartum resulted in increased urinary excretion
of 17-KS and ovarian enlargement.221,222,223,224 TABLE 8. Causes of Fetal and Maternal Virilization in Pregnancy
Drugs Dilantin
Danazol*
Progestogens*
Stilbestrol* (large doses)
Ovarian Lesions Arrhenoblastomas*
Luteoma of pregnancy*
Krukenberg tumor*
Mucinous cystadenoma
Leydig's cell tumor
Lipoid cell tumor
Granulosa-theca-cell tumor
Dermoid Cysts
Hyperreactio luteinalis
Polycystic ovarian syndrome
Adrenal Lesions Virilizing adenoma*
Virilizing carcinoma*
Aldosterone producing tumor*
*Lesions and drugs producing fetal virilization
Thyroid Diseases Thyroid disorders in pregnancy present a unique opportunity for health
care professionals to use a team approach that has successfully improved
the care of diabetic women. Obstetricians, endocrinologists, perinatologists, and
anesthesiologists are often called upon to interact in
the management of complex thyroid problems. In addition to changes in
thyroid function tests, hypermetabolic symptoms often seen in pregnancy
present a challenge in the diagnosis of thyroid disease during pregnancy. Thyroid hormone concentrations, both total thyroxine (TT4) and total triiodothyronine (TT3), increase during pregnancy as the result of both an elevation in thyroxine-binding
globulin (TBG) secreted by the liver and a reduced peripheral
TBG degradation rate.225 In spite of these changes in total hormone concentration, the serum free
fractions of both T4 and T3 remain within normal limits. However, there is a slight decrease in concentration
with progression of pregnancy. The serum levels of TSH, produced
by the pituitary gland, have a tendency to decrease in the first
trimester of pregnancy; they are undetected in 15% of normal pregnancies. With
progression of pregnancy, serum TSH levels return to normal
values.226 The suppression of serum TSH values in the first part of gestation is
secondary to the normal elevation in hCG, which has a weak thyroid stimulator
action on the TSH receptor. High levels of hCG, as in cases of
hydatidiform mole and hyperemesis gravidarum, may cause elevations in
serum T4 concentrations, as seen in thyrotoxicosis. Despite the limitations in the interpretation of serum TSH in the first
trimester, a second- or third-generation serum TSH test is the most practical
test to screen for thyroid dysfunction. High TSH values are consistent
with the diagnosis of primary hypothyroidism, whereas suppressed
values are suggestive of hyperthyroidism, with few exceptions (Fig. 3). In the presence of an abnormal serum TSH value, the determination of
FT4 or its equivalent, free thyroxine index (FT4I), will help in the assessment of thyroid function. A low TSH value and
high concentrations of FT4 or FT4I are diagnostic of hyperthyroidism; if the FT4 or FT4I is normal, a serum FT3 or FT3I determination is obtained. High values are diagnostic of hyperthyroidism, the
so-called T3-toxicosis syndrome, which is sometimes seen in patients with an autonomous
or “hot” thyroid nodule. Therefore, a few thyroid tests, if
properly ordered and interpreted, allow the physician to assess
thyroid function in pregnancy. 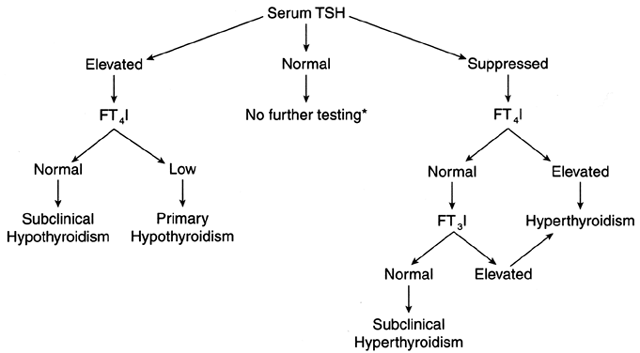 Fig. 3. Algorithm for the diagnosis of thyroid diseases. *, If there is clinical
suspicion of secondary hypothyroidism, a determination of FT4 I is indicated. In this situation the serum TSH is normal in the presence
of low FT4 I. FT4 I, free thyroxine index or its equivalent, free thyroxine; FT3 I, free triiodothyronine index or its equivalent, free triiodothyronine; TSH, thyroid-stimulating hormone. Fig. 3. Algorithm for the diagnosis of thyroid diseases. *, If there is clinical
suspicion of secondary hypothyroidism, a determination of FT4 I is indicated. In this situation the serum TSH is normal in the presence
of low FT4 I. FT4 I, free thyroxine index or its equivalent, free thyroxine; FT3 I, free triiodothyronine index or its equivalent, free triiodothyronine; TSH, thyroid-stimulating hormone.
|
In areas of iodine deficiency, goiter is commonly seen in pregnancy. However, in
the United States and other areas of the world with sufficient
iodine intake, the thyroid gland does not clinically increase in size
during pregnancy. Therefore, the detection of a goiter in pregnancy
is an abnormal finding that requires careful evaluation. One of the most
common causes of diffuse goiter is chronic autoimmune thyroiditis
or Hashimoto's thyroiditis. Most patients are euthyroid, and the
diagnosis is made by the determination of thyroid antibodies (antimicrosomal
or anti - thyroid peroxidase [anti-TPO]). Antibody concentration
decreases during pregnancy and increases in the postpartum
period. High values in the first trimester of pregnancy are predictors
of the syndrome of postpartum thyroid dysfunction. In some patients with a history of autoimmune thyroid disease, active or
inactive, the determination of TSH receptor antibodies is indicated
to assess the possibility of fetal or neonatal hyperthyroidism. These
antibodies, mainly of the immunoglobulin G (IgG) class, have different
functional activities-mainly stimulating or blocking antibodies on the
TSH receptor of the thyroid gland. When high titers are present in the
serum of pregnant women, they cross the placenta and may affect fetal
thyroid function. Thyroid-stimulating immunoglobulin (TSI), formerly
known as long-acting thyroid-stimulating antibody, may cause fetal or
neonatal hyperthyroidism.227,228 Thyroid-blocking antibodies present in women with chronic thyroiditis, particularly
those with atrophic thyroiditis, may produce neonatal hypothyroidism, a
condition that is transient because of the short half-life
of these antibodies.229 Titers of these antibodies decrease with progression of the pregnancy. This
could explain, in the case of Graves' disease, the decrease in antithyroid
drug requirement in many cases of hyperthyroidism in the second
half of pregnancy. A TSH receptor antibody value five times greater
than normal is considered predictive of neonatal or fetal hyperthyroidism. The
test is relatively expensive and should be ordered only in
very special circumstances (Table 9). The ideal time to order the test is between 32 and 34 weeks' gestation
because of the gradual decrease in titer concentration with progression
of pregnancy. TABLE 9. Indications for Serum Thyrotropin Receptor Antibody Determination
in Pregnant Women
Graves' disease Fetal or neonatal hyperthyroidism in previous pregnancies
Active disease, on treatment with antithyroid drugs
Euthyroid, postablation, in the presence of: Fetal tachycardia
Intrauterine growth retardation
Incidental fetal goiter on ultrasound
Chronic thyroiditis without goiter
Incidental fetal goiter on ultrasound
Infant born with congenital hypothyroidism
Maternal - Placental - Fetal Interactions In general it has been accepted that thyroid hormones do not cross the
placenta. However, recent studies in animals and some in humans have shown
some placental passage of thyroid hormones from mother to fetus in
the first few weeks of pregnancy, suggesting an important role of thyroid
hormones in embryogenesis.230 Some transplacental transfer from mother to fetus at the end of pregnancy
has been shown in cases of congenital hypothyroidism, although the
amount of hormone was not sufficient to normalize serum TSH in the neonate.231 Maternal TSH does not cross the placenta. TRH does cross the placental
barrier, but the physiologic significance is unknown. TRH has been injected
in mothers to accelerate fetal lung maturation in premature infants.232 Methimazole and propylthiouracil (PTU) (drugs used for the treatment of
hyperthyroidism) cross the placenta. If given in large doses, they may
produce fetal goiter and hypothyroidism.233 Iodine therapy is contraindicated in pregnancy because iodine is accumulated
by the fetal thyroid and induces goiter and hypothyroidism.234 Hyperthyroidism Hyperthyroidism is diagnosed in pregnancy in about 0.1% to 0.4% of patients.235,236 In most cases the etiology is Graves' disease; other causes are uncommon (Table 10). In the authors' experience, single toxic adenoma and multinodular toxic
goiter are seen in less than 10% of cases, and subacute thyroiditis
is rarely seen during gestation. Increased secretion of hCG may cause
chemical hyperthyroidism, as in cases of hydatidiform mole and hyperemesis
gravidarum.237,238 TABLE 10. Causes of Hyperthyroidism in Pregnancy
Graves' disease*
Multinodular toxic goiter
Toxic adenoma
Subacute thyroiditis
Iatrogenic hyperthyroidism
Hydatidiform mole
Hyperemesis gravidarum
*Accounts for 85% to 90% of all cases
Transient Hyperthyroidism of Hyperemesis Gravidarum In cases of severe hyperemesis gravidarum, thyroid tests in the hyperthyroid
range are not uncommon. The degree of thyroid abnormalities is directly
related to the severity of vomiting and weight loss. In the study
by Goodwin and associates,237 67 patients were evaluated. In those with severe vomiting, weight loss
of at least 5 kg, and significant dehydration, liver and electrolyte
function abnormalities were often found. These patients had significant
elevations in FT4 levels with suppression of serum TSH values. Those with lesser degrees
of hyperemesis had less severe thyroid function abnormalities. The presentation
of transient hyperthyroidism of hyperemesis gravidarum (THHG) is
characterized by severe nausea and vomiting requiring hospitalization (sometimes
repeated hospitalizations) for intravenous hydration. Weight
loss of at least 5 kg, ketonuria, and hypokalemia are common findings. FT4 levels are elevated, sometimes two to three times the normal values. FT3 is also elevated, but not as much as FT4. Serum TSH measured by a sensitive assay is consistently suppressed. In
spite of the significant biochemical hyperthyroidism, signs and symptoms
of hypermetabolism are mild or absent. Patients may complain of mild
palpitations, but heat intolerance, perspiration, and proximal muscle
weakness are extremely rare. On physical examination, ophthalmopathy
and goiter are absent, a mild tremor of the outstretched fingers is
occasionally seen, and tachycardia may be present, mainly a result of
dehydration. Significant in the medical history is the lack of hyperthyroid
symptoms before conception, which is relevant because patients
with Graves' disease diagnosed for the first time during gestation give
symptoms antedating the pregnancy. Normalization of hyperthyroxinemia
parallels the improvement in vomiting and weight gain, with most cases
resolving spontaneously in 2 to 10 weeks. However, suppressed serum
TSH may last for a few more weeks (Fig. 4). No antithyroid medication is needed; furthermore, because of the severity
of the vomiting, drug therapy is poorly tolerated. In one study
in which antithyroid medication was used, the outcome did not differ from
that of a similar group of patients receiving no antithyroid medication.239 In most cases, total resolution occurred by midgestation, although in
some series persistence of hyperthyroidism beyond 20 weeks' gestation
was reported in 15% to 25% of cases.240,241 Occasionally, severe vomiting and hyperthyroidism require parenteral nutrition. 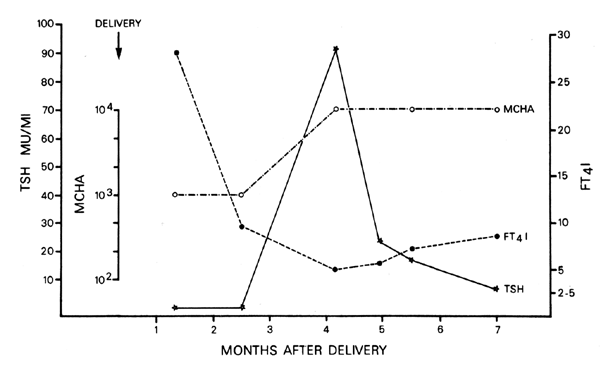 Fig. 4. Transient hyperthyroidism and hypothyroidism in the postpartum period in
a patient with chronic autoimmune thyroiditis with spontaneous recovery. TSH, thyroid-stimulating hormone; MCHA, microsomal hemagglutination
antibodies; FT4 I, free thyroxine index.(Mestman JH: Management of thyroid disease in pregnancy. In Berkowitz RL (ed): Clinics
of Perinatology. Philadelphia, WB Saunders, 1980) Fig. 4. Transient hyperthyroidism and hypothyroidism in the postpartum period in
a patient with chronic autoimmune thyroiditis with spontaneous recovery. TSH, thyroid-stimulating hormone; MCHA, microsomal hemagglutination
antibodies; FT4 I, free thyroxine index.(Mestman JH: Management of thyroid disease in pregnancy. In Berkowitz RL (ed): Clinics
of Perinatology. Philadelphia, WB Saunders, 1980)
|
The diagnosis of THHG is suspected when there is a sudden onset of nausea
and vomiting in the first part of pregnancy in a patient without hypermetabolic
symptoms antedating pregnancy, when there is absence of goiter, when
there are no other symptoms or signs of tissue thyrotoxicosis, and
when there are negative thyroid antibodies, which are markers
of autoimmune thyroid disease.242 Sometimes the differential diagnosis is difficult to make because vomiting
may be a presenting symptom of hyperthyroidism due to Graves' disease.243 The cause of hyperthyroidism in patients with hyperemesis gravidarum is
still controversial. The most likely explanation is high levels of hCG, a
known stimulator of the TSH receptor. There is a significant, albeit
weak, correlation between the degree of thyroid stimulation and total
hCG levels in normal and hyperemetic women.244 It seems likely, however, that certain hCG fractions may be more important
than the total hCG as thyroid stimulators. The thyroid-stimulating
activity of early pregnancy and of molar gestations correlates best
with the percentage of basic, partially desialylated hCG in serum.245 Although the true incidence of this entity is unknown, a higher prevalence
in Asian women has been reported.246 The diagnosis should be considered in pregnant women with severe vomiting, no
clinical manifestations of Graves' disease, and biochemical evidence
of hyperthyroidism in early pregnancy. The vomiting has to be persistent
and severe with significant weight loss, because most women
with morning sickness of pregnancy have normal thyroid function tests.247 Graves' Disease Graves' disease is the most common cause of hyperthyroidism in pregnancy, accounting
for over 85% of all cases. The natural course of the disease
is characterized by an exacerbation of symptoms in the first trimester
and during the postpartum period and an amelioration of symptoms
in the second half of pregnancy. When Graves' disease is properly treated, the
outcome for mother and fetus is good; however, untreated or
poorly controlled disease may be catastrophic for both the woman and the
neonate.248,249,250,251,252 In most patients whose diagnosis is made for the first time during pregnancy, symptoms
antedate conception. The clinical diagnosis of thyrotoxicosis
may be difficult during gestation because many hypermetabolic
symptoms are commonly seen in normal pregnancy, such as mild palpitations
and a heart rate of 90 to 100 beats per minute, mild heat intolerance, and
warm skin. There are a few important clues to guide the physician
toward the diagnosis of hyperthyroidism: presence of goiter, ophthalmopathy, proximal
muscle weakness, tachycardia with a pulse rate of
over 100 beats per minute, and weight loss or an inability to gain weight
in spite of a good appetite. Occasionally the patient is seen for
the first time in congestive heart failure. In patients with toxemia, the
physician should suspect hyperthyroidism when there is systolic hypertension
and an inappropriately low diastolic blood pressure, a wide
pulse pressure. Classical symptoms of hyperthyroidism include nervousness, increased sweating, increased
appetite, heat intolerance, insomnia, proximal muscle
weakness, irritability, changes in personality, frequent bowel movements, a
decreased tolerance to exercise (sometimes manifested as shortness
of breath), pruritus, and weight loss. Not all symptoms are present
in a given patient; the physician should be aware of subtle complaints, particularly
in the presence of weight loss or the inability to gain
weight. As mentioned above, differentiating Graves' disease from THHG
in the first trimester of pregnancy may present a real challenge to
the physician. On physical examination, the thyroid gland is enlarged in almost every
patient with Graves' disease. Indeed, the absence of a goiter makes the
diagnosis very unlikely. The gland is diffusely enlarged (between two
and six times its normal size) and varies from soft to firm; sometimes
it is irregular to palpation, with one lobe more prominent than the
other one. A thrill may be felt or a bruit may be heard; these are indications
of hyperdynamic circulation. Examination of the eyes may reveal
obvious ophthalmopathy, but in most cases exophthalmos is absent or
mild, with one eye slightly more prominent than the other one. Stare
is common, as well as injection or edema of the conjunctiva. Pretibial
myxedema is rare; it is seen in less than 10% of women. A hyperdynamic
heart and a loud systolic murmur are common findings. Proximal muscle
weakness, fine tremor, and hyperkinetic symptoms are seen often. The
skin is warm and moist, and palmar erythema is accentuated. Cardiovascular manifestations include left ventricular dysfunction. Although
these changes are reversible, they may persist for several weeks
after euthyroidism is achieved. In one study,253 a reduction in peripheral vascular resistance and a high cardiac output
were still present despite normalization of thyroxine levels. This is
an important finding with significant clinical implications. Left ventricular
decompensation in hyperthyroid pregnant women occurs in the
presence of superimposed preeclampsia, at the time of delivery, or with
intercurrent complications such as anemia or infection. Careful monitoring
of fluid administration is imperative in these situations. Thyroid
crisis or storm is rarely reported in pregnancy or the postpartum
period.254 The chemical diagnosis of hyperthyroidism presents no difficulties in most
cases. FT4 determination or calculation of the FT4I (using TT4 levels and a test for assessment of thyroxine-binding globulin, such as
resin T3 uptake [RT3U]) are standard tests in most clinical laboratories and give results
in 24 to 48 hours. Almost every patient with Graves' disease has an
elevated FT4 concentration. Serum TSH should be measured with a sensitive assay; this
should be ordered at the time of the free T4 determination. A suppressed TSH value in the presence of a high free T4 or FT4 index confirms the diagnosis of hyperthyroidism.255 It must be kept in mind, however, that a suppressed serum TSH level is
present in about 15% of normal pregnant women in the first trimester
of pregnancy.226 In some unusual situations, the serum FT4 is in the upper limit of normal or is slightly elevated, in which case
the determination of free T3 and the free T3 index will confirm the diagnosis of hyperthyroidism. Thyroid peroxidase
antibodies (antithyroid peroxidase) or thyroid antimicrosomal antibodies, markers
of thyroid autoimmune disease, are elevated in most patients
with Graves' disease. Determination of these antibodies is indicated
for patients in whom the etiology of the hyperthyroidism is in doubt. Treatment of hyperthyroidism is essential to prevent maternal, fetal, and
neonatal complications. The goal of treatment is to normalize the thyroid
tests with the minimum amount of antithyroid medication. Antithyroid
medications crossing the placenta may affect fetal thyroid function; patients
should be monitored at regular intervals, and the amount
of medication should be adjusted to keep the FT4 in the upper limits of normal (9.5 to 12). For this purpose, thyroid tests
should be performed every 2 weeks at the beginning of treatment and
every 2 to 4 weeks when euthyroid is achieved. In patients with small
goiters and a short duration of symptoms who are kept euthyroid on
minimal amounts of antithyroid medication, the drug can be discontinued
during the last few weeks of pregnancy. It is not recommended to discontinue
antithyroid therapy before 32 weeks' gestation because relapses
can occur. In the United States, the two antithyroid drugs available
are PTU and methimazole (Tapazole). Both drugs are effective in controlling
the disease; to my knowledge, there are no reports that show PTU
to be superior to Tapazole. Furthermore, in a recent report comparing
both drugs in a group of pregnant hyperthyroid women, euthyroidism
was achieved in both groups with equivalent amounts of drugs and in the
same period of time.236 Neonatal events did not differ between groups. Aplasia cutis, an unusual
scalp lesion reported in a small group of patients taking methimazole,256,257,258 is not a contraindication for use of this drug in pregnancy. The initial
recommended dose of PTU is 200 to 400 mg/d and the dose of methimazole
is 20 to 40 mg/d divided into two daily doses.259 This regimen is preferable for outpatient treatment to avoid poor compliance. At
initiation of therapy, however, PTU 50 to 150 mg should be
given every 8 hours. Very seldom are high doses needed, and indeed there
is no evidence that higher doses are therapeutically more effective. In
most patients, clinical improvement is seen in 2 to 6 weeks, and
an improvement in thyroid tests is seen in 2 to 4 weeks, with normalization
into chemical euthyroidism in 3 to 7 weeks.236 Resistance to drug therapy is very unusual; when it occurs it is most
likely due to poor compliance.260 Once clinical improvement occurs (mainly weight gain and a reduction in
tachycardia), the dose of antithyroid medication can be reduced by half
of the initial dose. The amount of medication is adjusted every few
weeks according to the clinical response and the results of the thyroid
tests. The goal is to keep the FT4 in the upper limit of normal (between 9.5 and 12) with the minimum amount
of medication. If there is exacerbation of symptoms or worsening of
the thyroid tests, the amount of antithyroid medication is doubled. The
main concern of maternal drug therapy is the potential side effects
on the fetus, mainly goiter and hypothyroidism. In most studies this
is prevented by using doses of PTU no greater than 200 mg and doses of
methimazole no greater than 20 mg in the last few weeks of gestation.261 However, small elevations in serum TSH in the neonate have been reported, even
with smaller doses of antithyroid drugs.262 Furthermore, in one study, cord blood FT4 was not related to the antithyroid dose.263 There is no evidence in the literature that transient elevations in serum
TSH in the neonate affect the long-term mental development of the
child.264 Side effects of antithyroid drugs occur in 3% to 5% of treated patients.265 Patients should be advised to discontinue medication and contact their
physician in case of pruritus or skin rash. This is the most common complication
and resolves by switching to the other antithyroid drug; in
general, this occurs 2 to 6 weeks after initiation of therapy. Other
side effects much less common are migratory polyarthritis, lupuslike
syndrome, and cholestatic jaundice. Agranulocytosis, a serious but unusual
complication, has been reported in 1 of 300 patients receiving an
antithyroid drug.266 It is manifested by fever, malaise, gingivitis, and sore throat. It occurs
in the first 12 weeks of therapy and appears to be related to the
amount of medication.267 Patients should be made aware of the symptoms and advised to obtain a
leukocyte count. Although some recommend a routine blood count in patients
on antithyroid therapy, it does not appear to be indicated because
granulocytopenia or agranulocytosis may appear without warning. At the time of the diagnosis and in very symptomatic patients, β-adrenergic
blocking agents are very effective in controlling hyperdynamic
symptoms after a few days of therapy. Propranolol 20 to 40 mg every 6 hours
or atenolol 25 to 50 mg daily is used until antithyroid drugs
control the hyperthyroidism, which may take between 4 and 8 weeks.236 Subtotal thyroidectomy in pregnancy is effective in controlling the disease, but
the indications for surgical treatment are few: allergy to both
antithyroid drugs,268 some cases of very large goiters, and the exceptional case of resistance
to drug therapy. Noncompliance could be another indication, although
it does not solve the problem of noncompliance. Therapy with 131I is contraindicated in pregnancy because it produces fetal hypothyroidism
when given after 10 weeks' gestation.269 A pregnancy test is mandatory in any woman of childbearing age before
a therapeutic dose of 131I is administered. Iodine crosses the placenta and may produce fetal goiter and hypothyroidism234; therefore, its use is contraindicated in pregnancy. However, it has been
used in small amounts (6 to 40 mg/d) in a group of pregnant Japanese
women with mild hyperthyroidism.270 An elevation in serum TSH was observed in 2 of 35 newborns; however, the
mothers were slightly hyperthyroid at the time of delivery. In spite
of this observation, iodine therapy is not indicated for the treatment
of hyperthyroidism in pregnancy. Excessive amounts of antithyroid drug have induced fetal hypothyroidism
and goiter. The diagnosis of goiter has been made by ultrasonography, which
shows hyperextension of the neck and goiter. A few cases of hypothyroidism
have been confirmed by measuring thyroxine and TSH in fetal
blood obtained by cordocentesis.271,272,273 Treatment with intra-amniotic injection of levothyroxine resulted in resolution
of the fetal goiter.272 Breastfeeding should be permitted if the daily dose of PTU is less than 150 to 200 mg/d
or the dose of methimazole is less than 10 mg/d. It is
prudent to give the total dose in divided doses after each feeding; the
infant should be followed with thyroid tests.274 In one study, PTU was given to mothers whose infants had elevated TSH; serum
TSH normalized even with continuation of PTU therapy by the mothers.275 Significant maternal and perinatal morbidity and mortality were reported
in early studies of pregnancies complicated by hyperthyroidism.276,277 In recent publications, however, there has been a significant decrease
in the incidence of maternal and fetal complications directly related
to maternal control of hyperthyroidism.278,279,280 Maternal complications include preterm delivery, placenta abruptio, pregnancy-induced
hypertension (PIH), and miscarriage. PIH or operative
delivery can trigger congestive heart failure in untreated women or women
treated for a short period of time.281 Thyroid storm has rarely been reported in pregnancy.254 Fetal and neonatal complications are also related to maternal control
of hyperthyroidism: intrauterine growth retardation, prematurity, stillbirth, and
neonatal morbidity are the most common complications. Mitsuda
and colleagues251 correlate the risk of delivering a small-for-gestational-age infant with
maternal thyrotoxicosis lasting for more than 30 weeks of pregnancy, Graves' disease
lasting for 10 years or more, and onset of Graves' disease
before age 20. Momotani and Ito282 reported a 25.7% rate of spontaneous abortions and a 14.9% rate of premature
delivery in mothers hyperthyroid at the time of conception; these
rates were 12.8% and 9.5%, respectively, in euthyroid mothers. This
is an important and practical aspect in the management of hyperthyroid
women of childbearing age; they should be advised about potential complications
and birth control during the hyperthyroid period of the disease. In
this regard, hormonal contraception is not contraindicated. Congenital
malformations are not increased in infants of hyperthyroid
mothers.283 However, in one such study, a 6% incidence of congenital malformations
was reported in mothers who were hyperthyroid at the time of conception.284 These findings need confirmation. Fetal well-being assessment with ultrasonography, a nonstress test, and
a biophysical profile is indicated for women in poor metabolic control
or when fetal tachycardia and/or intrauterine growth retardation is
noted. Neonatal Hyperthyroidism Neonatal hyperthyroidism is rare; it occurs in less than 1% of infants
born to mothers with Graves' disease. Thyroid-stimulating antibodies to
the TSH receptor (when present in high concentrations in maternal serum) cross
the placental barrier, stimulate the fetal thyroid gland, and
may produce fetal hyperthyroidism.285 If the mother is being treated with antithyroid medication, the fetus
is also being treated during gestation and therefore will present no symptoms
of hyperthyroidism. However, the protective effect of the antithyroid
drug is lost after delivery, and the neonate develops clinical
hyperthyroidism a few days after birth. A high titer of this TSH receptor
antibody (fivefold increase over baseline) in the third trimester
of pregnancy is a predictor of neonatal hyperthyroidism. If neonatal
hyperthyroidism is not recognized and treated properly, neonatal mortality
could be as high as 30%. Because the half-life of the receptor is
only a few weeks, complete resolution of neonatal hyperthyroidism is
the rule. Fetal Hyperthyroidism In mothers with a history of Graves' disease who were previously treated
with ablation therapy, concentrations of thyroid-stimulating antibodies
may remain elevated in spite of maternal euthyroidism. As mentioned
earlier, when these antibodies are present in high concentrations and
cross the placenta, they may induce fetal hyperthyroidism; this is characterized
by fetal tachycardia and intrauterine growth retardation.286,287 The diagnosis can be confirmed by measuring thyroid hormone levels in
cord blood obtained by cordocentesis. Treatment consists of antithyroid
medication given to the mother. Fetal tachycardia resolution and normal
fetal growth are indications of a good therapeutic response. Hypothyroidism Overt hypothyroidism in pregnancy is rare; only a few series of patients
have been reported.288,289,290 Subclinical hypothyroidism is more often encountered because hypothyroid
women on thyroid therapy often need an increase in the dose of levothyroxine
after conception.291,292 In two screening studies performed in the first half of pregnancy, the
incidence of serum TSH above normal was 2.5% in one study293 and 0.19% in another study from Japan.294 The two most common causes of primary hypothyroidism are post-thyroid
ablation therapy, surgical or 131I treatment, and autoimmune thyroiditis (Hashimoto's thyroiditis). Original
studies reported a high incidence of congenital malformations, perinatal
mortality, and impaired mental and somatic development in
infants of hypothyroid women.295 However, recent reports showed no increased incidence of congenital malformations, and
perinatal mortality was only slightly increased.288,289,290 The incidence of PIH was 21% in 60 patients with overt hypothyroidism (Table 11). Low birth weight was reported in 16.6% of infants. In one series289 there was an increase in postpartum hemorrhage, placenta abruptio, and
anemia. In the series from the Los Angeles County/USC Medical Center
there was only one case of postpartum hemorrhage in 34 women with overt
hypothyroidism.288,290 TABLE 11. Maternal and Neonatal Complications of Hypothyroidism in Pregnancy
| No. of Patients With | No. of Patients With |
| Overt Hypothyroidism | Subclinical Hypothyroidism |
| 16288 | 23289 | 11290 | Total 60 (%) | 12288 | 45289 | Total 57 (%) |
Pregnancy-induced hypertension | 7 | 5 | 1 | 13 (21) | 2 | 7 | 9 (15) |
Placenta abruptio | 3 | 0 | 0 | 3 (5) | 0 | 0 | 0 |
Postpartum hemorrhage | 3 | 1 | 0 | 4 (6.6) | 2 | 0 | 2 (3.5) |
Stillbirths | 2 | 1 | 1 | 4 (6.6) | 1‡ | 0 | 1 (1.7) |
Congenital malformations | 0 | 1 | 1 | 2 (3.3) | 0 | 0 | |
Low birthweight | 5* | 5† | 0 | 10 (16.6) | 1*§ | 4 | 5 (8.7) |
Anemia (HCT < 26%) | 5 | 0 | 0 | 5 (8.3) | 0 | 1 | 1 (1.7) |
Data from References 288,289,290.
*LBW < 2000 g.
†LBW < 2500 g.
‡ Congenital syphilis.
§ Class F diabetes.
HCT, hematocrit; LBW, low birth weight.
In patients with subclinical hypothyroidism, the incidence of PIH was 15%290; in the report by Davis and associates,289 2 out of 12 women with subclinical hypothyroidism developed postpartum
hemorrhage. Psychologic evaluation of children born to mothers with overt
hypothyroidism showed no impairment.288,296 Lui and co-workers296 examined a group of eight children at ages 4 and 10; their mothers were
hypothyroid in the first trimester of pregnancy. The children's
IQs were not different from those of their siblings, who were born when
the mothers were euthyroid. The spectrum of women with hypothyroidism in pregnancy includes the following: Women with overt hypothyroidism diagnosed for the first time during pregnancy
Women who discontinue thyroid therapy at the time of conception because
of poor medical advice or because of the misconception that thyroid medication
affects the fetus
Women on thyroid therapy who require larger doses in pregnancy
Women diagnosed for the first time with subclinical hypothyroidism
Most patients with subclinical hypothyroidism are asymptomatic. Patients
with overt hypothyroidism may complain of tiredness, cold intolerance, fatigue, muscle
cramps, constipation, and deepening of the voice. On
physical examination the skin is dry and cold, deep tendon reflexes
are delayed, bradycardia may be detected, and there may be periorbital
edema. The diagnosis of hypothyroidism is confirmed by the determination of serum
TSH and FT4 or FT4I. In patients with overt hypothyroidism, the FT4 is low and the serum TSH is elevated. The degree of chemical thyroid abnormalities
varies with the severity of the clinical symptoms; however, there
is not always a good correlation between clinical and chemical
parameters. In the author's series,290 the mean TSH value was 89.7 + 86.2 mU/L, with a mean FT4I of 2.1 + 1.5. Subclinical hypothyroidism is characterized by a normal
FT4 or FT4I and an elevation of serum TSH. Serum thyroid antibodies (thyroid peroxidase
antibodies) are elevated in patients with autoimmune thyroiditis. Levothyroxine is the drug of choice in the treatment of hypothyroidism. In
view of the complications mentioned above, it is important to normalize
the thyroid test as soon as possible. An initial dose of 0.150 mg
of levothyroxine is well tolerated by most patients. In patients with
severe hypothyroidism, there is a delay in the normalization of serum
TSH, but normal serum FT4 values are achieved in the first 2 weeks of therapy. The maintaining dose
required for most patients is 0.125 to 0.250 mg of levothyroxine per
day. Higher doses may be required in patients who have had total thyroidectomy
for thyroid carcinoma, because the goal in these cases is
suppression of serum TSH. Patients on thyroid therapy before conception should have their TSH checked
on the first visit and the amount of levothyroxine adjusted accordingly. The
serum TSH should be repeated between 20 and 24 weeks and between 28 and 32 weeks. Immediately after delivery, they should return
to their prepregnancy dosage. Ferrous sulfate, when given simultaneously
with thyroxine, may reduce the efficacy of thyroid hormone. The interaction
of these drugs is caused most likely by the binding of iron
to thyroxine. These drugs should be ingested 2 hours apart.297 Single Nodule of the Thyroid Gland The chances for a single or solitary thyroid nodule to be malignant are
between 5% and 30%.298 This risk is dependent on known risk factors, previous radiation therapy
to the upper body, rapid growth of a painless nodule, patient age, and
family history of thyroid cancer. There is a paucity of information
in the literature regarding management and timing of the workup in the
presence of thyroid nodularity.299 In general, it is recommended that elective surgery be avoided after 24 weeks' gestation
because of the potential risk of premature delivery. In
a select group of patients operated on during or in the few months
after delivery, the incidence of thyroid cancer was reported to be near 40%.300,301 In both studies, fine-needle aspiration biopsy (FNAB) of the thyroid nodule
was performed before surgery; the findings were consistent with
papillary carcinoma or highly suspicious lesions. Careful examination of the neck enables the physician to define and characterize
the lesion. In addition to the size of the nodule, consistency, tenderness, fixation
to the skin, and presence of metastasis should
be noted. A hard, painless nodule measuring more than 2 cm in diameter
is suspicious of malignancy. The following approach is recommended
at the LA-USC Medical Center (Los Angeles)301: - A suppressed serum TSH may indicate the presence of an autonomous nodule, which
rarely is malignant.302 In such a case, thyroid tests are performed to rule out hyperthyroidism. It
should be kept in mind that serum TSH is suppressed in 13% of normal
first-trimester pregnancies.56
- If the serum TSH is within normal limits, the next step is ultrasonography, which
will distinguish a solid from a cystic lesion.
- In the presence of a solid lesion less than 2 cm or a cystic lesion less
than 4 cm in diameter, observation with or without thyroxine suppression
therapy is recommended. If there is an increase in the size of the
lesion or if cervical adenopathy is present, an FNAB of the lesion is
indicated.
- If ultrasound shows a solid or mixed lesion greater than 2 cm or a cystic
nodule greater than 4 cm, the diagnostic approach differs according
to gestational age.
- Before 20 weeks' gestation, an FNAB is performed. The most important component
of the FNAB is the cytopathologist's interpretation. A specific
diagnosis is required: malignant, benign, follicular lesion, or
inadequate specimen. In the latter, a repeat FNAB is recommended.
- If the lesion is malignant, surgery is indicated. For follicular lesions, the
decision about surgery is a personal one, because there is a 15% to 20% chance
that the lesion is malignant. A similar approach is used
with HÜrthle cell lesions.
- For lesions diagnosed after 24 weeks' gestation, the FNAB may be postponed
until after delivery, unless there is a strong suspicion of malignancy. Suppressive
therapy with thyroxine to prevent further growth of
the lesion, although believed to be controversial by some,298 is the recommendation of the author's group. If there is further
growth of the lesion in spite of suppression therapy, FNAB is recommended.
- For lesions diagnosed between 20 and 24 weeks of gestation, the decision
to wait until after delivery or to complete the workup is made by the
patient and her physician. The patient's anxiety and fear about
having a potentially malignant lesion should be considered when the advice
is given. Most malignant lesions of the thyroid gland are slow growing, and
the long-term prognosis is good in most patients.
Postpartum Thyroid Dysfunction Thyroid dysfunction, both hyperthyroidism and hypothyroidism, is recognized
with increasing frequency in the postpartum period, within 12 months
of delivery. It occurs in 5% to 10% of all women in the postpartum
period.303,304 Most of the cases are due to intrinsic thyroid disease, and a few cases
are due to hypothalamic or pituitary lesions (Table 12). Patients with autoimmune thyroid disease, chronic thyroiditis, and Graves' disease
are most often affected. TABLE 12. Postpartum Thyroid Dysfunction
Chronic thyroiditis Transient hyperthyroidism (low RAIU)
Transient hypothyroidism
Permanent hypothyroidism
Graves' disease Exacerbation of hyperthyroidism (high RAIU)
Transient hyperthyroidism of chronic thyroiditis (low RAIU)
Hypothalamic-pituitary disease Sheehan's syndrome
Lymphocytic hypophysitis
RAIU, radioactive iodine uptake.
The clinical diagnosis is not always obvious, and the clinician should
be concerned about nonspecific symptoms such as fatigue, depression, palpitations, and
irritability in women after the birth of their child. Of
these, fatigue is the most common complaint.305 In about half of the cases, mild symptoms develop between 1 and 4 months
postpartum. On physical examination, a goiter is felt in most cases; it
is firm and nontender to palpation, and tachycardia may be detected. The
goiter may be discovered for the first time, or the patient may
notice an increase in the size of a previously diagnosed goiter. Thyroid
tests are in the hyperthyroid range, and thyroid antibody (antiperoxidase
thyroid antibody or antimicrosomal antibody) titers are elevated. This
is followed in a few months by hypothyroidism with spontaneous
recovery a few months later. The antibody titer has a tendency to increase
during this process and in most cases the size of the goiter increases. In
a few patients, permanent hypothyroidism may develop. About 50% of
patients, however, develop permanent hypothyroidism within 5 years
of the diagnosis of postpartum thyroiditis.306,307 In some cases the clinical symptoms resemble the syndrome of postpartum
depression. Indeed, thyroid antibodies have been reported to be found
more often in women with postpartum depression, but this is still a
controversial issue.308,309 Other patients have a different cause of postpartum thyroiditis, characterized
by an initial episode of hypothyroidism between 3 and 7 months
postpartum without the initial hyperthyroid phase. In others the initial
episode of hyperthyroidism is followed by a return to normal thyroid
function. Postpartum thyroid dysfunction can also occur in patients with a known
history of Graves' disease. It is common for women with Graves' disease
to have exacerbations of their symptoms in the few months postpartum.310 On the other hand, recurrence of hyperthyroidism may be secondary to a
concomitant episode of postpartum thyroiditis.311 The differential diagnosis in this situation is important because the
treatment is different. If not contraindicated, because of breastfeeding, a 4- or 24-hour
thyroid radioactive iodine uptake is helpful. It will
be very low in patients with postpartum thyroiditis, and it will be
elevated in patients with recurrent hyperthyroidism due to Graves' disease. When
it is due to recurrent Graves' disease, treatment with antithyroid
medications is indicated, or the physician may advise ablation
therapy. Hypothyroidism may be secondary to hypothalamic or pituitary lesions, such
as those seen in Sheehan's disease or lymphocytic hypophysitis (see “Pituitary
Diseases”). In such cases there are clinical
symptoms related to the deficiencies of other pituitary hormones. Because most cases of postpartum thyroid dysfunction recover spontaneously, treatment
is indicated for symptomatic patients. In the presence
of hyperthyroid symptoms, β-adrenergic blocking drugs (propranolol 20 to 40 mg
every 6 hours or atenolol 25 to 50 mg every 24 hours) are
effective in controlling the symptoms. For hypothyroid symptoms, small
amounts of levothyroxine (0.050 mg/d) will control symptoms, allowing
for a spontaneous recovery of thyroid function after discontinuation
of the drug. Postpartum thyroid dysfunction may recur in future pregnancies. There are some features that may predict the development of postpartum
thyroiditis: the presence of goiter, high titers of thyroid antibodies
in the first half of pregnancy, episodes of postpartum thyroiditis in
previous pregnancies, and a strong family history of autoimmune thyroid
disease.312 Women with insulin-dependent diabetes mellitus are at high risk of developing
postpartum thyroiditis. |
 Fig. 1. Serum calcium ( closed circles) and creatinine ( closed squares) levels during pregnancy and for 1 month after delivery in a woman with
hypoparathyroidism who was treated with vitamin D and calcium (see text). Stippled
area shows normal range. IV, intravenous.(Ficinski M, Mestman JH: Hyperparathyroidism in pregnancy. Endocr Prac, Sept/Oct 1996)
Fig. 1. Serum calcium ( closed circles) and creatinine ( closed squares) levels during pregnancy and for 1 month after delivery in a woman with
hypoparathyroidism who was treated with vitamin D and calcium (see text). Stippled
area shows normal range. IV, intravenous.(Ficinski M, Mestman JH: Hyperparathyroidism in pregnancy. Endocr Prac, Sept/Oct 1996)