Mechanical Pathophysiology of the Placenta
Authors
INTRODUCTION
If the fetus has a normal chromosome complement, a readable developmental program, then further normal development depends on normal nutrient availability (i.e., normal maternal vasculature) and normal anatomy for nutrient transport from the intervillous space to the conceptus proper (i.e., normal fetoplacental vasculature). Both of these features can be deformed or disrupted by mechanical stressors.
FETAL DEATH
Intrauterine fetal death is the ultimate failure in the mechanism of the fetoplacental unit. Causes of fetal demise differ with gestational age. In the first trimester, abnormal conceptus karyotype is the single most common cause of pregnancy loss. Fetal death due to aneuploidy (e.g., Turner's syndrome) is far less common in the second trimester, especially in the nonanomalous stillborn fetus. “Incompetent cervix” and the oft-related process of acute intra-amniotic infection are commonly associated with preterm delivery, whether due to premature membrane rupture1 or preterm labor.2 Both may cause fetal or neonatal death due to extreme prematurity. Fetal death from sepsis is uncommon, especially in low-risk (community hospital) settings. Placentas from fetal deaths due to the antiphospholipid antibody syndrome demonstrate the classic pathology of multifocal placental infarcts and uteroplacental thrombosis.3 This second-trimester histology is not common in first-trimester losses in women with this syndrome. Chronic inflammation (not thrombosis) is more common in the untreated early pregnancy losses of women subsequently diagnosed with antiphospholipid antibodies.4 In the last trimester, uteroplacental insufficiency, cord accidents, and diffuse chronic villitis are the most common placental correlates with fetal death. Long-standing placental damage (as in uteroplacental vascular insufficiency and severe chronic villitis) may chronically limit fetal nutrients before fetal decompensation and death. Abnormal growth (either low birth weight or a low weight/length ratio or “ponderal index”) in a karyotypically normal fetus should raise the clinical suspicion of chronic placental disease. The dividing line between antemortem and postmortem lesions is not always clear. The differences among the placentas of a dead fetus, a damaged newborn, and a healthy newborn may only be the extent or severity of any one histologic lesion, or the presence of multiple lesions.
Gross and histologic features of the placenta in fetal death
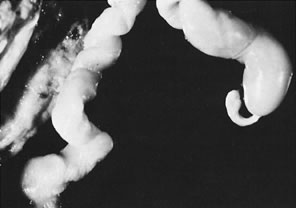
Gross umbilical cord torsion is most common in midtrimester fetal death, when the normally large amniotic fluid volume allows passive fetal rotation after death. Torsion after death may occur first at the attachment of the umbilical cord to the fetus and may reflect effects of passive fetal movement rotating the flaccid cord after death. Torsion at the insertion of cord to the chorionic plate and especially torsion in the midsection of the umbilical cord may both be clinically significant antemortem lesions (Fig. 1). Hemolysis of fetal erythrocytes causes discoloration of Wharton's jelly soon after fetal death.5 The discoloration of the cord can be mistaken for umbilical cord thrombosis. Cessation of fetoplacental blood flow affects the fetoplacental circulation, with secondary effects on villus stroma, villus trophoblast, and lastly the (maternal) uteroplacental circulation. Autolysis of the cord progresses from the fetus to the placenta.
|
Microscopically, the villous circulatory collapse and obliteration that follow fetal death are indistinguishable from vascular changes in liveborns after endothelial damage caused by cocaine-induced vasospasm, viral infection, fluctuating perfusion (hypoperfusion/reperfusion), or near-chronic villitis, infarcts, or abruptions. In stillbirth the circulatory pathology is generally more diffuse than in, for example, preeclampsia or fetal growth restriction. When the circulation to an area of villi stops, the villous stroma becomes densely fibrotic, the villi appear smaller, and the syncytiotrophoblast stains deep blue-black. With retention of the fetus for months after fetal demise (as in selective reduction of multiple gestations), the (maternal) intervillous blood space is obliterated and only ghost villi remain suspended in a shrunken and clotted intervillous space. Placental features can help time the duration of in utero retention after fetal demise. Shanklin and colleagues addressed this issue in the rabbit5 and the human.6 Genest7 and Genest and colleagues8, 9 characterized a wide range of both gross and microscopic placental and fetal features that allow calculation of the death–delivery interval. They also provided specificity, sensitivity, and positive predictive values for each feature. Unfortunately, gross and microscopic placental features provide more crude estimates of the death–delivery interval than patterns of visceral autolysis.5, 8, 9
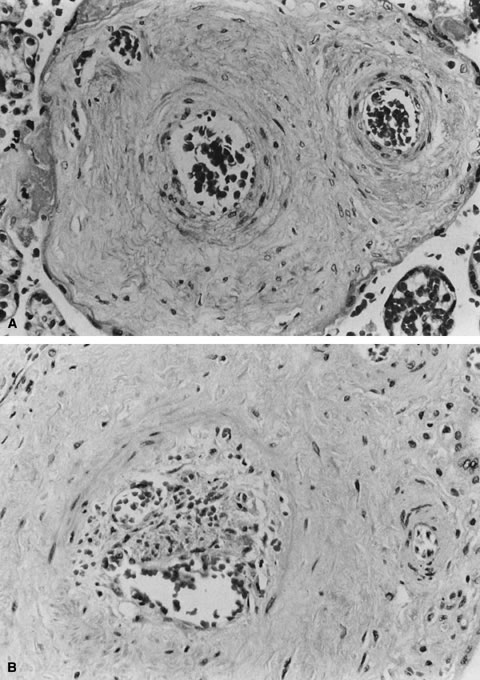
A specific type of villous vaso-obliteration was first identified in placentas from stillborn fetuses. This pattern of fetal red cell fragmentation and intravillous bleeding was termed hemorrhagic endovasculitis (HEV; Fig. 2).10 HEV is often not a true vasculitis and may develop after fetal death. Typical HEV is observed in in vitro organ cultures11 and in retained secundines. However, HEV is an important lesion in placentas of stillborn or liveborn fetuses. HEV is not universal in stillborn fetuses, and its histology may vary widely even when stillbirths occur at similar times before delivery. Therefore, it cannot be simply part of what happens to the circulation after death. In liveborn infants, HEV is associated with fetoplacental circulatory “instability.”12 It is also common adjacent to foci of chronic villitis, at the edge of infarcts and intervillous thrombi, and in infarcted chorioangiomas. HEV may be associated with clinical diagnoses of gestational hypertension and preeclampsia13, 14 and with elevated total numbers of fetal circulating nucleated erythrocytes.15 When HEV is seen in cases of fetal death with prolonged in utero retention, the lesions are often more uniform, as might be expected from a general effect of global cessation of circulation. When HEV is present and fetal death was close to delivery, antemortem fetoplacental circulatory instability may be suspected. Anecdotally, our experience is that HEV may be more common with umbilical cord accidents, where cord compression and hypoperfusion/reperfusion vascular injury (the paradigm of circulatory instability) would be a major process. HEV can recur, and can recur in placentas of a second fetal death, or of subsequent liveborn infants.13
|
HYDROPS FETALIS
Hydrops fetalis is the effect of compromised cardiocirculatory function. It is a final common pathway for a wide range of pathophysiologies, including increased capillary permeability to water and protein, decreased lymph flow, and possibly congestive heart failure and elevated blood volume.16 There is debate about whether congestive heart failure per se is a cause of hydrops. Hydrops is not always present with any cardiac anomaly, and when a cardiac anomaly accompanies hydrops fetalis, there may also be lymphatic obstruction or other fetal problems causing capillary injury and leak, or increased intrathoracic pressure obstructing venous return.17 Differences in the fetal microcirculation and lymphatic system, as compared with mature subjects, make the fetus more susceptible to interstitial fluid accumulation. Compared with adults, the fetus has greater capillary permeability, a more compliant interstitial compartment, and greater influence of venous pressures on lymphatic return. The balance between interstitial fluid production and removal is most commonly disrupted when cardiocirculatory function is impaired.18 Generalized hydrops in a nonanomalous fetus is most commonly due to fetal anemia. The three major causes of chronic fetal anemia are increased fetal erythrocyte destruction (e.g., Rh and blood group incompatibilities, some congenital viral syndromes), decreased erythrocyte production (e.g., some congenital viral infections), and hemoglobinopathy or fetoplacental hemorrhage.19, 20, 21, 22 This last may be caused by rupture of velamentous vessels or vasa previa, umbilical cord hemorrhage, or, in our experience most commonly, after any placental injury that can tear or bruise the villous capillaries. The most common cause of placental bruising, or villous stromal hemorrhage, is in our experience abruption. Fetal hydrops may also develop as a result of placental lesions (e.g., chorangioma or venous obstruction).
Once there is fetal anemia sufficient to cause capillary hypoxia, there is capillary leak of both protein and water.16, 21 The fetal and placental circulations are generally affected in parallel, and fetal hydrops is almost always reflected in placental “hydrops.” Placental findings in hydrops fetalis include variable villous dysmaturity (histology less mature than expected for gestational age), increased cytotrophoblast mitoses, thickened trophoblast basement membranes, increased fibrinoid necrosis, and hemosiderosis of the extraplacental membranes, villous macrophages, endothelia, and the trophoblast basement membrane (especially when the pathophysiology involves abruption and villous stromal hemorrhage). Increased amounts of both intravillous and extravillous calcification are seen with intrauterine cardiac failure,23 intrauterine fetal death, or when there is less fetal demand for calcium, as in osteogenesis imperfecta.
UMBILICAL CORD PATHOLOGY
The human umbilical cord must reflect some critical evolutionary compromise; otherwise, it is hard to see the advantages of having the fetal lifeline to its placental respiratory and nutritional source be merely two arteries and a single vein. These vessels are packaged in a cord up to 70-plus cm long that floats in a fluid space that becomes progressively smaller throughout gestation. Finally, this fluid space may contain substances potentially noxious to fetoplacental hemodynamics, such as cytokines and possibly meconium. That cord pathology is comparatively infrequent speaks to the cord's remarkable resilience.
The umbilical cord epithelium is continuous with amniotic epithelium and tightly adherent to the underlying Wharton's jelly. The cord has no vasa vasorum, lymphatics, or nerves. Mast cells in the compressible Wharton's jelly may protect against umbilical thrombosis. The Wharton's jelly cell population includes many histiocytes, cells that are facultative macrophages. After meconium exposure or cord trauma (e.g., cordocentesis), the cells may phagocytose debris and become pigment-laden.
The vascular muscle is a decussating helix of smooth muscle fibers. The umbilical vein has a well-developed internal elastic lamina; the arteries have elastic tissue only in the media. The umbilical arteries have been described to anastomose within the first 2–3 cm of cord proximal to the chorionic plate.24 Such an anastomosis would facilitate uniform placental perfusion. This Hyrtl's anastomosis is usually at the site of cord attachment to the chorionic disk, but it may be on the chorionic plate between smaller branches of the chorionic arteries. We dissected umbilical arteries in 69 consecutive term deliveries and found a variety of anastomotic patterns. In nine of the 69 (13%), the umbilical arteries completely fused to form a common channel. In 28 (41%), a transverse anastomosis was found within 2 cm of the chorionic insertion. Five cases (7%) showed an anastomosis on the chorionic plate distal to an umbilical arterial branch point. In six cases (9%), a visible anastomosis was not probe-patent.25 Any communication that allowed the potential for pressure and flow equilibration between the umbilical arteries would allow greater capacity for ventilation-perfusion matching, for the placenta to redistribute blood away from focal placental lesions to better-perfused areas. Variance in anastomotic patterns may explain the variability of fetal compromise when focal placental injury such as infarct and abruption occur. However, measurable differences between umbilical arterial Doppler waveforms are both uncommon and not clearly associated with perinatal compromise.26
Recently, the Doppler resistance index before and after this anastomosis was studied at a median gestational age of 33.1 weeks (range 25.5–40.3 weeks).26 Anastomosis blood flow was always unidirectional toward the umbilical artery with a lower resistance index. When the cord insertion was marginal or velamentous, umbilical arteries were more likely to be fused, but in all cases, the anastomoses served to equalize pressures between the umbilical arteries. The authors suggest that this provides a “safety valve” against arterial compression that permits persistent perfusion of the complete placental disk despite cord compression.27 The majority of vascular resistance lies in the placenta itself;28 maintenance of low umbilical arterial resistance is important for normal fetoplacental hemodynamics. After transit through the placental capillary bed, blood pressure has continued to drop so that by the time blood reaches the common umbilical vein, its flow is nonpulsatile.28 Pulsatile umbilical venous flow has been associated with poor pregnancy outcome,29 especially in the growth-restricted fetus.30 Umbilical blood flow tends to increase in proportion to fetal weight, about 100–130 mL/kg per minute between 14 and 28 weeks of gestation.28
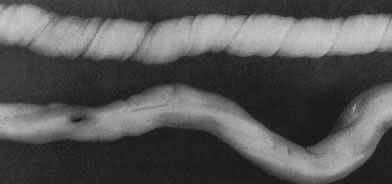
Cord length also increases with increasing fetal weight.31 Other factors associated with increased cord length include parity,32 the size of the uterine environment,33 increased fetal movement,33 male gender,33 and possibly genetics, as there may be an increased risk of recurrent long umbilical cords in women with a past pregnancy with a long umbilical cord.31 Decreased umbilical cord length is associated with decreased fetal movements from any cause dating from early in gestation, including Down syndrome, skeletal dysplasias, central nervous system lesions that impair fetal movement, amnion bands (in which fetal movement may be constrained after amnion rupture), and uterine structural malformations and multifetal gestations, in which the uterine environment is constrained or crowded.31 The association of oligohydramnios with a shorter cord is inconsistent,34 likely reflecting different times of onset and severity of oligohydramnios. Umbilical coiling (Fig. 3) may serve to enhance cord hemodynamics, as arterial pulsations transmitted to the vein may help pump blood back up the cord from the placental capillary bed. Both abnormally straight (uncoiled) cords and excessively coiled cords are associated with an increased risk of adverse perinatal outcome.35 Risk factors for abnormal vascular coiling were extremes of maternal age for hypercoiling, and obesity, gestational diabetes mellitus, and preeclampsia.35 An excessively long or coiled cord may be at increased risk for torsion. Passive rotation of the fetus around the flaccid cord after death may lead to cord twisting and, in the absence of an umbilical cord pulse, severe stricture. Careful gross and microscopic examination and detailed clinical correlation are necessary to rule out other causes of cord vulnerability to torsion during life (reduced intrauterine volume/oligohydramnios, abnormal cord insertion). To some degree, this diagnosis may be a diagnosis of exclusion in the stillborn fetus. This has been considered a sporadic cause of fetal compromise, but a genetic predisposition has been recently suggested, based on a familial cluster of three subsequent pregnancies with recurrent cord torsion and recurrent fetal death.36
|
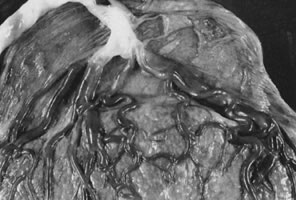
Velamentous and marginal insertions of the umbilical cord (Fig. 4) also carry potential fetal risk. When the cord is inserted on the membranes (velamentous insertion), the chorionic arteries and vein must travel in the chorion laeve to reach the definitive placental disk (or an accessory lobe). Membranous vessels must accompany velamentous cord insertion but may be found with marginally inserted cords, with accessory lobes, or with completely normal chorionic placentation. Vasa previa describes the case when these membranous blood vessels lie over the cervix, between the baby and the cervix. Even dilatation of the lower uterine segment may put these vessels under mechanical stress, and membrane rupture can be an immediately lethal event to the fetus if the vasa previa are large-caliber vessels. A recent series of 18 suspected cases of vasa previa included ten velamentous cords and three cases each of bilobed placentas, accessory lobes, and marginal cord insertion. Two of the 18 cases ended with fetal death; the remaining infants did well with minor complications of prematurity.37 With rupture of a large-caliber vessel, fetal exsanguination may occur within 3 minutes. Velamentous vessels not seen over the os may still rupture, although this is far less common. Independent of the risks of vessel rupture, malposition of the umbilical cord insertion may compromise fetal well-being. The placental disk is approximately twice as thick when the maternal intervillous blood space is fully distended by normal maternal intervillous blood flow than after delivery. Even after delivery, the chorionic plate is a “springy” flexible structure, cushioning the vessels coursing in it. By contrast, velamentous vessels are adjacent to the rigid myometrial wall, a structure without a great deal of “give.” Velamentous vessels are much more susceptible to compression and other types of mechanical trauma. In twin pregnancies, the velamentous cord insertion is more common in the smaller of the siblings, and the donor of the pair in cases of twin-transfusion.38 Sections through traumatized but unruptured velamentous vessels may show necrotic smooth muscle cells, mural thrombi, and mixed vasculitis. A marginally inserted umbilical cord may also be at risk because of the mechanics of the angle at which the extraplacental membranes join the chorionic disk. Intra-amniotic pressure maintains this angle open. Once the membranes rupture, this angle may buckle, destabilizing the umbilical cord. We have observed cases of acute fetal decompensation at spontaneous or artificial membrane rupture, or after internal cephalic version with a marginal umbilical cord insertion, especially when the umbilical cord is short. Mechanical stress can compromise umbilical cord vessels when there is segmental absence of Wharton's jelly, focal deficiencies of muscular wall,39 or furcate insertion. In furcate insertion, the vessels split before their insertion into the chorionic plate and may have some investment with Wharton's jelly. However, vessels in the furcate insertion are more susceptible to compression, trauma, or laceration. Fetal structural defects associated with velamentous cord insertions are typically due to deformation of a normally formed part, probably as a result of intrauterine molding.40
|
One of the risks of compression and vascular injury is thrombosis. Thanks to the cushioning of Wharton's jelly, potentially the protective, anticoagulant effect of cord mast cells, and the normal helical arrangement of the vessels, cord thrombosis due to compression or mechanical trauma is uncommon, even with extensive velamentous vessels. Spontaneous cord thrombosis is very rare. Two recent case reports described umbilical venous thrombosis with intrauterine fetal death, one after abrupt onset of fetal heart rate decelerations with delivery 14 minutes later of a nonresuscitable infant.41 Predisposing factors to cord thrombosis are unclear, but cord coiling may be an important protective mechanism. In a study,42 37% of cases with overcoiled cords and 29% of undercoiled cords were cases of fetal demise. The authors raised the possibility that abnormal cord coiling might carry risks of neurologic sequelae to survivors. Obviously, further study of this possibility is needed. One of us (C.M.S.) has seen two cases of spontaneous cord thrombosis associated with neonatal stroke, and both were in infants of mothers who were at least heterozygous for the factor V Leiden mutation. The recommendation, based on pathology, was that the infant and/or father be genotyped for this mutation. Vern and colleagues43 have described avascular villi, an obliteration of the fetoplacental vasculature in the placental microcirculation, in association with fetoplacental heterozygosity for the factor V Leiden mutation. It may be reasonable to obtain careful cardiovascular pedigrees from mother and father in cases of umbilical cord thrombosis, because the potential for hereditable thrombophilia would bear significantly on recurrence risk assessment.
Umbilical cord edema develops as a result of any forces that promote movement of water out of the umbilical vessels and into Wharton's jelly. In any vascular system, an increase in intravascular pressure would lead to fluid extravasation. Therefore, it is not surprising to find umbilical edema in the vicinity of umbilical cord knots. Umbilical edema is also commonly seen in association with intra-amniotic infection. We speculate that intra-amniotic inflammatory cytokines affect umbilical epithelium permeability and/or umbilical vascular pressure to promote umbilical cord edema. In support of the latter hypothesis, we have observed an increase in the umbilical systolic–diastolic ratio in cases with histologic umbilical vasculitis.44 Umbilical edema is also common in infants of a diabetic mother and may reflect the hyperdynamic state of these overgrown, plethoric infants.
Absence of one artery (Fig. 5) may be due to failure of formation or to regression with a residual calcified remnant.45, 46 Embryonic remnants (most common near the fetal end of the cord) are of little clinical significance.47 Umbilical cord length in part reflects fetal growth and in utero fetal activity.31 Umbilical cord pathology and some guides to clinical correlation are presented in Table 1.
|
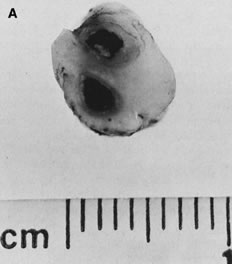
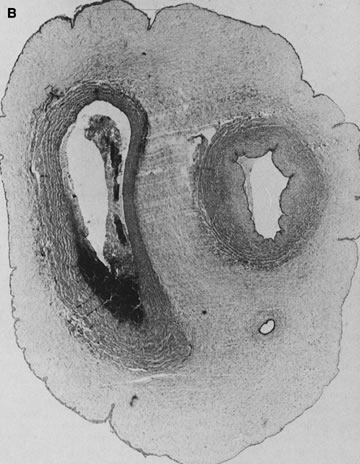
Table 1. Umbilical cord abnormalities
Abnormality | Incidence | Significance |
Single umbilical artery | 1% | 20% congenital malformations, IUGR, small placenta, twins, trisomy 18, maternal diabetes |
| 2.5% stillborn |
|
Supernumerary vessels | Rare | Section taken within 3 cm of placental insertion, conjoined twins, remnant (vitelline vessel) |
Short cords | ? | <32 cm, decreased fetal movement and neurodevelopmental abn, musculoskeletal abn, difficult delivery, congenital malformations, twins, breech presentation |
Long cords | ? | >100 cm, increased fetal movement, thrombosis, entanglements, torsions |
Edema | 2.7% | Hydrops fetalis, preeclampsia, acute chorioamnionitis |
Thin cord | 0.2% | IUGR, fetal malformations including prolapse and compression |
Omphalomesenteric duct | 1.5% | Near surface of cord, muscle in wall, intestinal-type mucosa, rarely ulceration of gastric mucosa, ? association with Meckel's diverticulum or intestinal atresia |
Allantoic duct | 15% | Located between the arteries, connective tissue wall, transitional-type epithelium, ? patent urachus or cyst |
Vitelline vessels | 7% | Usually central, usually capillary-like, rarely with muscular wall, origin of hematoma or hemangioma |
Teratoma | Rare | Benign |
Hematoma | 2% | Rupture of umbilical vein, iatrogenic most common, short cords |
True knots | 0.5–1% | Rarely tighten during labor, rarely cause of in utero hypoxia |
False knots | Frequent | Vascular redundancies, varicosities, rare thrombosis |
Thrombosis | 0.7% | Associated with perinatal death, more common in vein, reported to be more significant in artery(ies), congenital hypercoagulable states, acute chorioamnionitis, funisitis, vigorous fetal motor activity, commonly associated with true knot |
Strictures | ? | Focal deficiency of Wharton's jelly, rarely due to “amniotic bands” |
Torsion | ? | Exaggerated spiraling, including fetal movement; most occur after fetal demise |
Decreased spiraling | ? | Single uterine artery, decreased fetal movement |
Cord rupture | 2–4% | Short cord, velamentous or marginal insertion |
Marginal insertion | 5–7% | Compression of vessels, thrombosis, rupture, twins |
Velamentous insertion | 0.3–2% | Compression of vessels, thrombosis, avulsion of cord, rupture of vasa previa, twins, diabetes, congenital malformation |
Cord prolapse | 2–4% | Long cords, multiple gestations |
Funisitis | ? | Fetal reaction to infection or cord compression, margination, angiitis, funisitis (neutrophils in Wharton's jelly), rare before 26 weeks, candidiasis, plaques on surface |
Necrotizing funisitis | Rare | Syphilis, herpes, toxoplasmosis, often associated with villitis and chorioamnionitis |
Calcification | Rare | Necrotic inflammatory debris muscle fibers, etiology known |
Muscle degeneration | Rare (livebirths) | <1% of meconium-stained placentas, autolysis, begins at fetal end of cord |
| Common | After fetal demise |
abn, abnormalities; IUGR, intrauterine growth retardation.
MECONIUM
Recently meconium has been proposed to cause both umbilical vasospasm (through chemical irritation) and degeneration of vascular smooth muscle cells, with the sequela of fetal/neonatal cerebral damage due to abnormal fetal cerebral perfusion.48, 49 The proponents of this theory are vehement in its defense. In our experience there is little histologic evidence except in extremely rare cases to support that meconium gains access to the fetoplacental circulation by diffusion and that meconium circulates in the fetus to cause widespread vasospasm and ischemic fetal damage. Myocyte damage has been linked to meconium exposure,50 and this has also been proposed to have a deleterious effect on umbilical vascular function. Because even segmental complete absence of umbilical vascular smooth muscle may not be associated with any increase in complications,39 it is difficult to know what (if any) impact on vascular function could be expected from the degenerative loss of the most peripheral of vascular smooth muscle cells (those most commonly affected by any meconium-associated changes). More study is required to clarify the effects of meconium on umbilical and fetoplacental vascular function, as in many cases in our joint experience with cord injury associated with meconium, the newborn is clinically recognized to be compromised.
Clinically, meconium passage means different things at different gestational ages. As the fetal gut matures, meconium moves ever closer to the distal colon and rectum. Not surprisingly, meconium passage at term is common in otherwise uncomplicated deliveries. At term, the fetal stressor that initiates the release of meconium located at or just above the anal sphincter may be trivial and unsustained, such as cord compressions that would occur during normal uterine contractions in labor. At term, meconium passage in the absence of the stress of labor or before membrane rupture (e.g., at diagnostic amniocentesis) may be more significant. Passage of meconium in a preterm fetus requires a stress sufficient to move meconium over a greater colonic distance and may imply a greater severity and/or duration of stress than that required for meconium passage at term. Passage of meconium, especially in the midtrimester, may be associated with acute ascending infections, and with Listeria hematogenous infection. It has been speculated that intra-amniotic infection may cause fetal gastroenteritis and diarrhea. Meconium passage is uncommon before 32 weeks, and one of us (C.M.S.) routinely obtains iron stains to rule out hemosiderin in any cases with pigment deposition within membranes before or at 32 weeks' gestation.
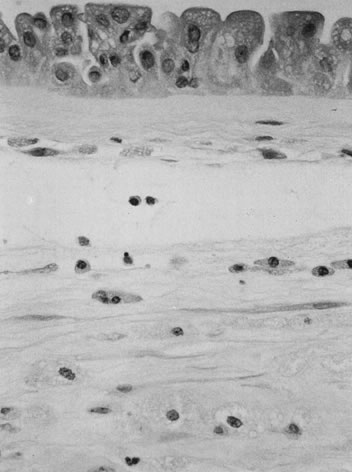
The initial histopathology of meconium passage is individual amnion epithelial cell necrosis, followed by amnion hyperplasia, pseudostratification, and vacuolation (Fig. 6), over an unknown time frame. Meconium is phagocytosed by macrophages in the amnion and chorion and eventually cleared from the amnion fluid. What time is required to clear meconium from the amniotic fluid and whether meconium is ever removed from cells of the extraplacental membranes are unknown.
|
EARLY AMNION RUPTURE SEQUENCE
This sequence occurs in its severe form in 1 in 25,000 and in its mild form in 1 in 10,000 livebirths. The early amnion rupture sequence is much more frequent in previable fetuses (estimated incidence as high as 1 in 53)51, 52 and is underdiagnosed in small or fragmented specimens. The sequence begins with amnion rupture (Fig. 7). As the amnion detaches, it pulls fibrous strands from the chorionic extraembryonic mesoderm. Any residual amnion strands attach to the insertion of the cord. The fibrous strands entangle body parts, including the umbilical cord,53 and may cause strangulation and amputation. If they are swallowed or aspirated, facial deformities (especially asymmetric clefting) may develop due to the pressure of the bands on delicate fetal tissues. The stripped chorion is more water-permeable than the amnion. Increased water loss from the amniotic fluid may lead to oligohydramnios and fetal compression. The denuded chorion is also more “sticky”; immature fetal skin (devoid of surface keratin layers) may adhere to it, tethering and tearing any surface of the body.54 Defects generated by this mechanism include neural tube defects, craniofacial clefts, primary body wall defects, caudal regression, and limb reduction defects.51, 55 Effects of amnion rupture vary with the time in gestation of rupture, mainly due to changes in the character of the fetal skin (increased keratinization), ossification of bony structures, and denser composition of fetal soft tissues, making them less likely to adhere to the chorion and more resilient to traction stress. Early rupture (earlier than 45 days) carries a high risk of cerebral and limb abnormalities.52 Later rupture is usually associated with limb abnormalities and a lower incidence of central nervous system abnormalities.52 Limb anomalies tend to be irregular, asymmetric, and distal, with proximal structures being relatively preserved. Constricting bands and limb hypoplasia without amputation may be seen. These findings may be difficult to distinguish from those of the abdominal wall/absent umbilical cord syndrome. Amnion rupture is generally considered to be a sporadic event, but there is one report of familial recurrence.56
|
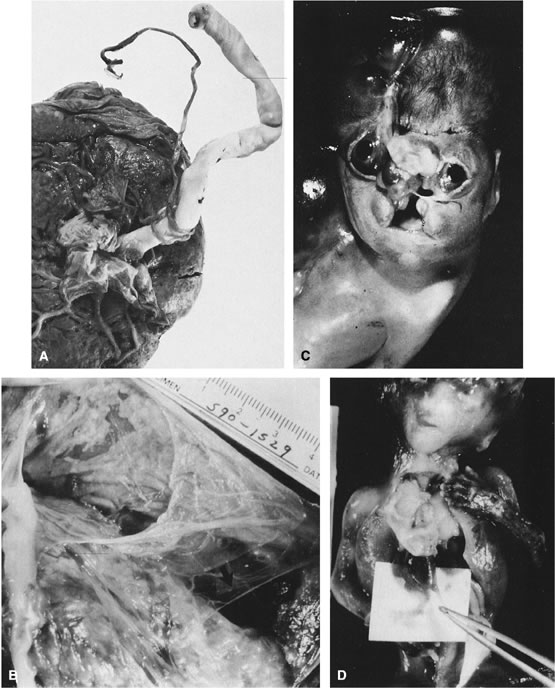
Limb body wall complex (LBWC) is probably the most severe manifestation of the amnion disruption complex. It consists of two of the three following defects: exencephaly or encephalocele with facial clefts, thoraco- and/or abdominoschisis, and limb defects. LBWC is complicated by rupture of the unsupported amnion. The placental disk is usually devoid of amnion. Amnion is found covering only one side of the umbilical cord and is contiguous with the abdominal wall defect. Bands of mesodermal connective tissue may be found attached to the chorionic plate, similar to those seen with amnion rupture. The abdominal contents develop within the extraembryonic celomic cavity, not within the amniotic cavity.57 Recently, four cases of LBWC were described, and none had craniofacial defects. Three had abdominal wall defects with abdominal eventration, cloacal exstrophy, absent external genitalia, abnormal internal genitalia, scoliosis, and lower limb defects. One fetus also had ectopia cordis and a severe reduction defect of the left arm, but normal colon, anus, bladder, genitalia, and lower limbs. All four had a short, malformed umbilical cord, incompletely covered with amnion. Umbilical vessels traveled in a short sheet of amnion that connected the skin margin of the defect of the placenta.58 The primary defect is thought to be related to abnormal body stalk development58 and is an anomaly therefore of early embryonic organization and development rather than postembryonic disruption, as proposed for the amnion rupture sequence.57
OLIGOHYDRAMNIOS
Oligohydramnios may result from either decreased fetal production of amniotic fluid or by increased loss of amniotic fluid. Examples of the former are renal agenesis or lower urinary tract obstructions. Examples of the latter include the amnion rupture sequence or chronic amniotic fluid leak. In a series of nonanomalous infants, both infants with oligohydramnios and polyhydramnios had lower birth weight, shorter gestational age, shorter umbilical cord, and higher parental ages.59 Abnormal amniotic fluid volume, either high or low, may mark chronic intrauterine pathology.
The hallmark lesion of chronic, severe oligohydramnios is amnion nodosum (Fig. 8). Small, finely granular or greasy amnion plaques that are easily scraped off are seen on the attached plate and on free/reflected membranes or less often umbilical cord. Because the nodules are embedded squamous debris from the amniotic fluid, more intra-amniotic squamous debris or its greater concentration may speed the genesis of amnion nodosum. Amnion nodosum may develop more readily after vernix has accumulated (by 32 weeks) or in the context of decreased amniotic fluid volume. Likewise, any reduction in intra-amniotic volume would increase mechanical trauma to the amnion. The histology of amnion epithelial cells changes from the thin, flat, simple squamous epithelium of the early midtrimester to the tall columnar epithelium; pseudostratification, apical blebbing, and nuclear pleomorphism are common at term. Amnion cell degeneration with or without necrosis, or finding the histology of term amnion in a midtrimester case may suggest decreased amniotic fluid volume. The mechanical effects of chronically reduced amniotic fluid volume and/or pressure on the placenta may include circumvallate membrane insertion, and their effects on fetal pulmonary and musculoskeletal development are well known.
|
ABNORMAL PLACENTATION
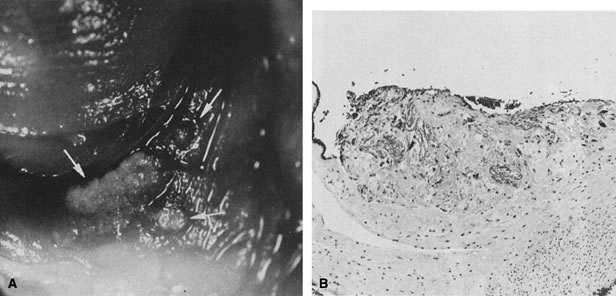
Early in gestation, the entire circumference of the placental sphere is covered by villi. As the gestational sac expands and the abembryonic chorion bulges into the uterine cavity, the blood supply to the villi becomes attenuated. The abembryonic villi atrophy, eventually forming the bald chorion. If villi other than those that will form part of the chorion frondosum can obtain a blood supply from, for example, the opposite uterine wall, they can continue to grow and arborize, developing into an accessory lobe. Predisposing conditions include uterine crowding (e.g., multiple gestations or septated uteri) or implantation in the lower uterine segment, which fails to expand as readily as the fundus. An accessory lobe is generally not associated with any maternal or fetal compromise except when an accessory lobe causes partial placenta previa or vasa previa (in which case fetal exsanguination may occur) or when an accessory lobe is left behind in the uterus after delivery (leading to maternal postpartum complications of retained secundines). A more extensive variant of this process is placenta membranacea (Fig. 9), in which there is more extensive or complete persistence of villi over the entire sphere of the chorion. This condition carries significant risks because it obligates a placenta previa and may be associated with significant acute or chronic maternal and/or fetal bleeding in the third trimester.
|
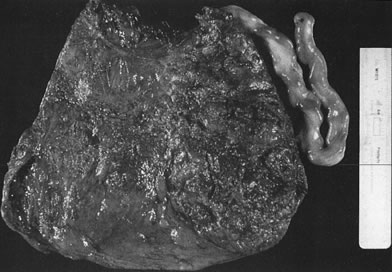
Placenta previa, implantation of the placenta partially or completely over the cervical os, is the most common cause of antepartum hemorrhage. Other causes are shown in Table 2. Predisposing factors for abnormal site of implantation include maternal age, parity, anatomic uterine abnormality (leiomyoma, septate uterus), or previous uterine surgery (curettage, cesarean section, or myomectomy).60 The delivered placenta previa may show basal decidual hemosiderin, old infarct with or without retroplacental hematoma, or focal villous atrophy in the area of previa. Similar changes can often be observed at the margin of a placenta in which a midtrimester placenta previa has resolved on serial ultrasonographic examinations. The resolution of a placenta previa may involve the necrosis of the placental area overlying the cervical os, with the former area of previa visible only as an area of atrophy or infarct.
Table 2. Abnormal placental implantation resulting in maternal hemorrhage
Placental Findings | Incidence | Significance |
Placenta accreta (increta, percreta) absent decidua | 0.4% | AP and PP hemorrhage Uterine rupture Maternal and fetal death |
Placenta previa | 5–28% 0.3–3% | Early pregnancy, common Term delivery, less common May be partial or complete AP hemorrhage Maternal and fetal death |
Low-lying placenta | ? | Placenta accreta PP hemorrhage |
Abruptio placentae | 0.17–3.75% | Fetal mortality (30–65%) AP bleeding Tonic uterine contractions 33% are associated with aretroplacental hematoma Placental infarct |
Circumvallate | 1–5% | AP bleeding Premature delivery Associated with abruptio |
PP, postpartum; AP, antepartum.
Placenta creta is a generic term for abnormal implantation of placental villi onto (accreta), into (increta), or through (percreta) the uterine myometrium. Placenta percreta may even extend into the bladder. The basic defect is the paucity or absence of decidualized endometrium. Without the normal plane of cleavage at the decidual interface, placental delivery can be difficult, and remnants of the placenta and basal plate can be left in the uterine cavity, with the resultant risks of maternal hemorrhage and infection. Because the lower segment, endocervix, and cornu of the fallopian tube do not transform into decidua, by definition, implantation in these sites results in placenta creta. Placenta creta and placenta previa frequently occur together.60 The most common site of placenta creta is over a previous uterine scar, such as a previous cesarean section, dilatation and curettage, or myomectomy.
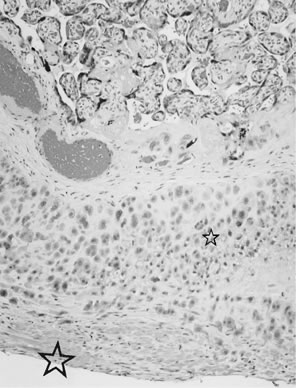
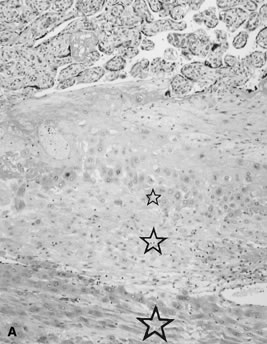
Placenta creta is a clinical diagnosis based on difficulty of delivery of the placenta. If the placenta is delivered and examined pathologically, areas of “incompleteness” of the maternal surface can be noted. The range of findings can extend from missing segments of the placenta to focal irregularities of the basal surface. At the edges of incomplete areas, histologic examination may yield smooth muscle (Fig. 10).61, 62 This may be the only confirmation of a diagnosis of placenta creta that can be made when only the placenta is submitted to pathology. If a postpartum curettage or hysterectomy specimen is available, the diagnosis is usually confirmed. Histologically, the lesion may be very focal, with absence of decidualized endometrium, and villi implanted directly onto the myometrial smooth muscle (Fig. 11). The invasive trophoblast can be distinguished from endometrial stromal cells by immunohistochemistry; trophoblasts are cytokeratin positive and endometrial cells are vimentin positive. In a set of 462 births at less than 32 weeks' gestation and 108 singleton uncomplicated births at more than 37 weeks' gestation, excluding stillbirth, multiple gestation, chronic hypertension, diabetes mellitus, and fetal congenital anomalies, 44 of 462 (9.5%) of the preterm placentas had basal myometrial fibers versus 0.9% (1/108) of term controls (p <0.001). Uteroplacental vessels with abnormal physiologic change were more frequent in cases with myometrial fibers (p <0.003), and placental weights were lighter (p <0.03). The incidence of basal myofibers was similar in preterm preeclampsia, premature membrane rupture, preterm labor, and nonhypertensive abruption.61 Experimental models suggest that cytotrophoblasts proliferate in response to hypoxia63, 64 and may also directly migrate in response to local oxygen tension.65 One response to local hypoxia may be deeper myometrial invasion. Most of the foci of basal myofibers in our data set were found in the immediate vicinity of a uteroplacental vessel with incomplete or absent physiologic conversion. Basal myofibers may indicate local placental hypoperfusion that has prompted local deeper cytotrophoblast invasion.
|
|
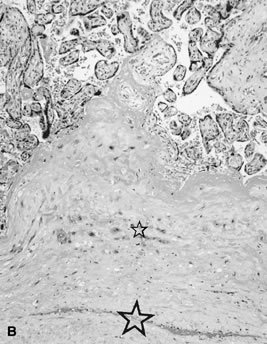
Circumvallate membrane insertion (Fig. 12) may result from abnormally deep uterine implantation. In some cases, circumvallate membrane insertion is believed to reflect the effects of a transient decrease in intra-amniotic pressure (e.g., a healed injury to the amnion and chorion). Decreased intra-amniotic pressure may cause the placental margins to buckle, entrapping decidua in a fold of the extraplacental membranes. If the leak reseals, intra-amniotic pressure will be restored and the placental margin restabilized. The entrapped ring of decidua folded onto the chorionic plate may be the only pathologic evidence of early amnion rupture. Circumvallate membrane insertion must be distinguished from circummarginate membrane insertion, which is not associated with abnormal fetal outcome.
|
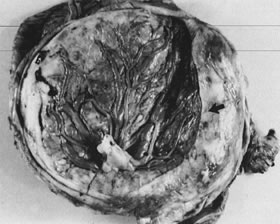
MULTIPLE GESTATIONS
Multiple gestations carry a higher risk to both maternal and fetal well-being. Maternal risks are most often the result of increased pregnancy hormone levels (e.g., gestational diabetes mellitus), expanded blood volume (anemia), uterine overdistention (e.g., preterm labor and postpartum hemorrhage), and increased placental mass (some cases of preeclampsia).66, 67, 68, 69, 70, 71, 72, 73 Despite advances in neonatal care, the perinatal death rate in twins is high, with the risk varying according to the type of placentation. Although only 1% of twin placentas are monochorionic monoamniotic, they account for 54% of the deaths. Monochorionic diamniotic placentation accounts for 30% of twins and 27% of total deaths. Therefore, monochorionic placentation accounts for 31% of twins but 71% of twin deaths.67, 71
Twinning is the most common naturally occurring type of multiple gestation. The incidence of monozygotic twinning (identical or monovular twins) is stable worldwide (3–5 per 1000 births). The etiology is unknown, but it may be a teratogenic event. There is a 1% risk of recurrence. Familial twinning is dizygous due to an inherited propensity to multiple ovulations. The rate of twinning has increased from 1 in 55 livebirths in 1970 to 1 in 43 in 1993. Because this increase is due to delayed child-bearing (with increased twinning rates at older maternal ages) and more widespread use of fertility drugs and assisted reproductive technologies, dizygous and monozygous twins are likely to become ever more common.66 Rarely, dizygotic twins may be conceived at nearly the same time (superfecundation) or be conceived at different times (superfetation), and possibly by different fathers. Twenty per cent of dichorionic twins are monozygous. Zygosity can be determined in 55–60% of twins by a combination of placental examination and knowledge of the infants' sexes.73 All monochorionic twins are monozygous, and all opposite-sexed twins are dizygotic. Slightly more than half of dichorionic placentas have like-sex babies; of these, additional testing will reveal one third to be monozygous.73 Whether dichorionic placentas are separate or fused does not predict zygosity, because this is determined by the proximity of implantation of the two blastocysts. Placentation with monozygous twins is determined by the time of zygote or blastocyst division. Late division (occurring after postovulation day 13) results in conjoined twins.72 Conjoined twins are a rare malformation of monochorionic monoamniotic twinning, occurring in 1 in 50,000 to 1 in 100,000 livebirths. They may have umbilical cords that are branched or fused, often with an abnormal number of vessels (usually two veins and three arteries).
The placenta should be labeled by the delivering clinician or labor room staff, indicating which umbilical cord was associated with twin A (first-born) and which with twin B (second-born). One clamp is commonly placed on the cord of twin A and two on the cord of twin B. Twin A is usually nearest the birth canal and more susceptible to ascending (transcervical) infection. Twin B is more likely to be growth-retarded, have congenital malformations, or suffer from perinatal asphyxia than twin A. Long-term, twin B does not fare worse than twin A if all other circumstances are taken into consideration (e.g., fetal growth retardation, single umbilical artery, and congenital malformations).67
Monochorionic twins share a single placental disk (Figs. 13 and 14). However, the placental zones demarcated by the dividing membranes and the vascular distribution associated with each cord may not be identical. We recommend recording the overall dimensions and the sizes of the respective vascular distributions, either as two diameters or as a percentage of the total disk surface. Separate or partly fused placentas are examined identically to singleton placentas. Gross examination of the dividing membrane usually reveals the number of amnions and chorions. For a single placental disk, a membrane roll from the dividing membrane or its junction on the placental surface (the “T zone”) documents the gross impression. Rarely, the dividing membranes rupture before birth, converting a diamniotic placenta into a functionally monoamniotic sac.74 In dichorionic diamniotic separate placentas, the weight is not double that expected for a singleton at the same gestational age. The mean weight for twin placentas is ~1.69 that expected for a singleton at the same gestational age. No significant differences are noted for dichorionic versus monochorionic twins.75 Cord length is shorter in twins, thought to be due in part to the confined space in the uterus.76
|
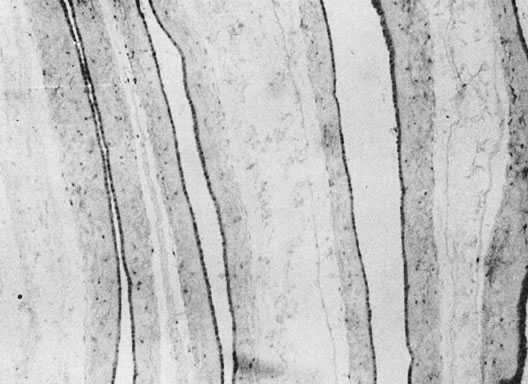
|

Superficial vascular anastomoses are seen in many chorionic placentas and are easy to identify by injecting air or milk into the vessels (Fig. 15). Arteries always cross over veins, and in each placental area an artery and vein should be paired. Deep anastomoses, the so-called third circulation, most commonly involve an outflow artery-to-vein anastomosis across a capillary bed, balanced by a vein-to-artery inflow anastomosis. The caliber of the anastomosing vessel reflects the volume transfused through this vascular “short circuit.” Therefore, documentation of the calibers of anastomosing vessels can help determine the nature of the twin vascular sharing. When unbalanced, the outflow vessel may be large, calcified, and not paired with a vessel returning to the same cord. More accurate identification of these anastomoses requires cumbersome injection studies. For such studies, the placenta must be submitted intact, unfixed, and at room temperature. The blood clots must be flushed from the circulation. The chorionic vessels may then be injected with a water-soluble dye, milk, or radiopaque fluid.77 This technique is generally used only for research purposes and only exceptionally in routine pathology practice.
|
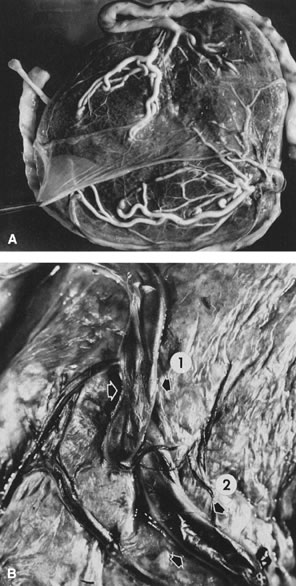
Fetal death (Figs. 16 and 17) is a common complication of multiple gestation. Cord accidents, including cord entanglement, underlie 50% of the fetal demises in single sac twins. Prolapse of the umbilical cord of twin B during delivery of twin A78 is more common in monoamniotic placentas but may occur in any type of placentation once the dividing membrane ruptures. During the first trimester, as many as 21% of twins may be lost.78 In early fetal losses, there may be no evidence of the fetus at the time of delivery (the “vanishing twin”).76 Fetal loss in the second or third trimester occurs in 0.5–6.8%.79 A fetus papyraceous forms after fetal death after the skeleton is well enough formed to retain its shape (generally 13 weeks' gestation). Radiologic confirmation of suspected fetus papyraceous will show skeletal remains. After the demise of the co-twin, the circulation of the dead twin loses vasomotor tone. The drop in blood pressure across anastomoses may cause hypovolemic/hypotensive ischemia and damage (including cerebral injury) to the surviving twin.80, 81, 82 Occasionally, both surviving twins each show cerebral injury of the hypoxic–ischemic type,83 suggesting that fluctuations in blood pressure or blood volume may result in cerebral necrosis without requiring the death of one twin or transfer of a blood-borne factor from one twin to the other. When one of a twin pair dies, the placenta of the dead twin is initially normal but will eventually atrophy due to hypoperfusion unless the living co-twin continues to perfuse its capillary bed through anastomoses.84, 85
|
|

Prematurity, acute chorioamnionitis, premature rupture of membranes, and fetal growth retardation of one or both twins are all more common in multifetal gestations and carry risk for immediate and long-term death and complications.85, 86 Twins follow the growth curves of singletons until the third trimester, when their growth rate slows. This may be due to uterine crowding, decreased placental reserve, or abnormal blood flow to one twin.87 The fetal growth retardation is usually asymmetric, with “head sparing.” Discordant growth of monozygous twins may be due to unequal division of cytoplasmic mass during the equal division of the genetic material or the twin transfusion syndrome. Discordant growth is seen almost as commonly in dizygous twins. Chronic villitis, intraplacental thrombi, fibrosis, villous hypovascularity, villous infarcts, and maternal uteroplacental vasculopathy are seen more commonly in the smaller dichorionic twin.88, 89 Congenital malformations occur in approximately 10% of monochorionic twins.90, 91 Single umbilical artery is seen in 2.5–4% of twins compared with approximately 1% in singletons. Single umbilical artery is seen in 20% of acardiac twins.92 Abnormal cord insertions are seen in 16% of all twins, compared with 1% in singletons. Marginal insertion is seen in 14% of monochorionic and 4% of dichorionic placentas. Velamentous insertion is seen in 9% of monochorionic and 5% of dichorionic placentas, with a higher incidence of fetal growth retardation and fetal compromise in the twin with velamentous cord insertion.38 There is a higher incidence of loss of chromosomal material and midline defects, including neural tube defects, facial clefts, tracheoesophageal fistulas, and sirenomelia.91
Although nearly all monochorionic placentas have vascular anastomoses, twin transfusion syndrome occurs in 7.5–30%.67, 71 Chronic twin transfusion syndrome occurs most often in monochorionic diamniotic placentas, infrequently in monochorionic monoamniotic placentas (thought to be due to nearly complete sharing of the placental substance), and rarely in dichorionic diamniotic fused placentas.93 Twin transfusion syndrome is the result of chronic unidirectional vascular steal from the artery of one twin (donor) through the “third circulation” to the other twin (recipient) (Fig. 18). When one twin's cord is inserted velamentously, the fetal vessels are adjacent to the rigid myometrium and not on the chorionic plate, and are more subject to compression. This twin is more likely to become the donor.38 The donor is growth-retarded and anemic, with delayed organ maturation and oligohydramnios.94, 95 The recipient becomes plethoric and is overgrown.95 Polyhydramnios and congestive heart failure due to volume overload, and polycythemia may occur.95 Cardiac dysfunction is more common in the recipient of the transfusion.96 The placenta reflects the fetal abnormalities.97 Acute twin transfusion syndrome may occur at the time of fetal demise, during delivery, or rarely in conjunction with chronic twin transfusion syndrome.98 The criteria for clinical diagnosis remain controversial, but twin transfusion syndrome can be considered when monochorionic twins have a more than 20% discordance in their weights and a more than 5-g/dL difference in hemoglobin levels.99 However, discordance of weight and hematocrit of equal or greater severity can occur in cases without twin transfusion syndrome.99 Recently both clinical100 and pathologic101 investigations have suggested that placental differences, rather than chronic transfusion, may underlie the fetal pathology considered classic for twin transfusion syndrome. However, intravascular flow volume and pressure are known to affect vascular differentiation.102 It is physiologically reasonable that differences in placental weight and vascular arborization are the effects of chronic transfusion (with chronic hyper- and hypovolemia of recipient and donor) rather than the cause that leads to twin transfusion syndrome. In a series studied under optimal physiologic conditions, cases with twin transfusion syndrome had significantly fewer anastomoses than those without. However, those anastomoses were significantly more likely to involve the deep (capillary) circulation in cases with twin transfusion syndrome.103 After repeated amniocentesis to reduce polyhydramnios, the placenta may show hemosiderin deposition.
|
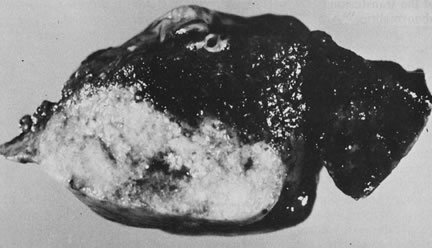
Acardia is a very rare malformation restricted to monozygotic twins of either monoamniotic or diamniotic placentation. Its occurrence is (under) estimated at 1 in 35,000 births.72, 104 Acardia is caused by reversal of blood flow, most commonly through superficial vascular anastomoses. The acardiac twin loses direct vascular connection with the placental circulation and receives its entire blood supply from the pump twin. Close proximity of the two umbilical cords on a common chorion and discordant development between the twins must be present for reversal of arterial blood flow to occur. Deoxygenated, low-pressure blood from the pump twin, which would normally return to the placenta, instead flows directly to the acardiac twin. The increased cardiac work of the viable twin may lead to significant morbidity in the survivor, including radiographically detectable pulmonary artery calcification in the absence of pulmonary valvular anomalies.105 The death rate of the pump twin is 50–75%, and of course 100% of acardiac fetuses die.106 Chromosomal abnormalities have been reported in more than 50% of the acardiac twins studied.104, 107 The acardiac twin may be quite amorphous, and a diagnosis of placental teratoma may be considered.108
Higher-order multiple births (triplets, quadruplets, and so forth) have become more common in recent years due to delayed childbearing, use of fertility drugs, and in vitro fertilization techniques. Naturally occurring higher multiple births are infrequent (triplets 1 in 10,000 births, quadruplets 1 in 100,000 births).109 Higher-order multiple births may be a combination of mono- and dizygous siblings (see Fig. 18). Evaluation of the placenta is done as described above, with particular attention paid to the number of chorions and amnions. Monochorionic placentation means monozygosity of those offspring, independent of other placental features. Reference values for placental weights from higher multiple pregnancies are now available.110
REFERENCES
Silverman RK, Wojtowycz M: Risk factors in premature rupture of membranes. Prim Care Update Ob Gyn 5: 181, 1998 |
|
Lang JM, Lieberman E, Cohen A: A comparison of risk factors for preterm labor and term small-for-gestational-age birth. Epidemiology 7: 369, 1996 |
|
Rai R, Regan L: Obstetric complications of antiphospholipid antibodies. Curr Opin Obstet Gynecol 9: 387, 1997 |
|
Salafia CM, Parke AL: Placental pathology in systemic lupus erythematosus and phospholipid antibody syndrome. Rheum Dis Clin North Am 23: 85, 1997 |
|
Shanklin DR, Cimino DA, Lamb TH: Fetal maceration: I. An experimental sequence in the rabbit. Am J Obstet Gynecol 88: 213, 1964 |
|
Shanklin DR: Fetal maceration: II. An analysis of 53 human stillborn infants. Am J Obstet Gynecol 88: 224, 1964 |
|
Genest DR: Estimating the time of death in stillborn fetuses: II. Histologic evaluation of the placenta; a study of 71 stillborns. Obstet Gynecol 80: 585, 1992 |
|
Genest DR, Singer DB: Estimating the time of death in stillborn fetuses: III. External fetal examination; a study of 86 stillborns. Obstet Gynecol 80: 593, 1992 |
|
Genest DR, Williams MA, Greene MF: Estimating the time of death in stillborn fetuses: I. Histologic evaluation of fetal organs; an autopsy study of 150 stillborns. Obstet Gynecol 80: 575, 1992 |
|
Sander CM. What's new in placental pathology. Pathol Annu 30:59, 1995 |
|
Silver MM, Yeger H, Lines LD: Hemorrhagic endovasculitis-like lesion induced in placental organ culture. Hum Pathol 19: 251, 1988 |
|
Shen-Schwarz S, Macpherson TA, Mueller-Heubach E: The clinical significance of hemorrhagic endovasculitis of the placenta. Am J Obstet Gynecol 159: 48, 1988 |
|
Sander CM, Gilliland D, Flynn MA, Swart-Hills LA: Risk factors for recurrence of hemorrhagic endovasculitis of the placenta. Obstet Gynecol 89: 569, 1997 |
|
Salafia CM, Pezzullo JC, Lopez-Zeno JA et al: Placental pathologic features of preterm preeclampsia. Am J Obstet Gynecol 173: 1097, 1995 |
|
Salafia CM, Ghidini A, Pezzullo JC, Rosenkrantz TS: Early neonatal nucleated erythrocyte counts in preterm deliveries: clinical and pathologic correlations. J Soc Gynecol Invest 4: 138, 1997 |
|
Phibbs R: Hydrops fetalis and other causes of neonatal ascites. In Pollin RA, Fox EE (eds): Fetal and Neonatal Physiology, Chap 13. Philadelphia, WB Saunders, 1992 |
|
McFadden DE, Taylor GP: Cardiac abnormalities and nonimmune hydrops fetalis: A coincidental not causal, relationship. Pediatr Pathol 9: 11, 1989 |
|
Apkon M: Pathophysiology of hydrops fetalis. Semin Perinatol 19: 437, 1995 |
|
Machin GA: Hydrops revisited: Literature review of 1414 cases published in the 1980s. Am J Med Genet 34: 366, 1989 |
|
Nakamura Y, Komatus Y, Yano H et al: Nonimmune hydrops fetalis: A clinicopathological study of 50 autopsy cases. Pediatr Pathol 7: 19, 1987 |
|
Nicolaides KH: Studies on fetal physiology and pathophysiology in Rhesus disease. Semin Perinatol 13: 328, 1989 |
|
Harman CR: Fetal monitoring in the alloimmunized pregnancy. Clin Perinatol 16: 691, 1989 |
|
McDermott M, Gillan JE: Trophoblast basement membrane haemosiderosis in the placental lesion of fetal artery thrombosis: A marker for disturbance of maternofetal transfer? Placenta 16: 171, 1995 |
|
Boyd JD, Hamilton WJ: Foetal blood vessels of the placenta. In The Human Placenta, Chap 14. Cambridge, England, W Heffer and Sons, 1970 |
|
Salafia CM, Divon NY, Mill JF et al: Umbilical artery anastamosis: revisiting an old axiom (abstr 189). Am J Obstet Gynecol 174 (suppl): 363A, 1996 |
|
Raio L, Ghezzi F, Di Naro E et al: The clinical significance of antenatal detection of discordant umbilical arteries. Obstet Gynecol 91: 86, 1998 |
|
Raio L, Ghezzi F, Di Naro E et al: In utero characterization of the blood flow in the Hyrtl anastomosis. Placenta 22: 597, 2001 |
|
Dawes G: The umbilical circulation. Am J Obstet Gynecol 84: 1634, 1962 |
|
Hofstaetter C, Dubiel M, Gudmundsson S: Two types of umbilical venous pulsations and outcome of high-risk pregnancy. Early Hum Dev 61: 111, 2001 |
|
Gramellini D, Piantelli G, Verrotti C et al: Doppler velocimetry and non-stress test in severe fetal growth restriction. Clin Exp Obstet Gynecol 28: 33, 2001 |
|
Baergen R, Malicki D, Behling C, Benirschke K: Morbidity, mortality, and placental pathology in excessively long umbilical cords: Retrospective study. Pediatr Dev Pathol 4: 144, 2001 |
|
Sornes T, Bakke T: Uterine size, parity and umbilical cord length. Acta Obstet Gynecol 68: 439, 1989 |
|
Naeye R: Umbilical cord length: clinical significance. J Pediatr 107: 278, 1985 |
|
Miller M, Jones M, Smith D: Tension: The basis of umbilical cord growth. J Pediatr 101: 844, 1982 |
|
Ezimokhai M, Rizk DE, Thomas L: Maternal risk factors for abnormal vascular coiling of the umbilical cord. Am J Perinatol 17: 441, 2000 |
|
Bakotic B, Boyd T, Poppiti R, Pflueger S: Recurrent umbilical cord torsion leading to fetal death in 3 subsequent pregnancies. Arch Pathol Lab Med 124: 1352, 2000 |
|
Lee W, Lee VL, Kirk JS et al: Vasa previa: Prenatal diagnosis, natural evolution, and clinical outcome. Obstet Gynecol 75: 572, 2000 |
|
Fries MH, Goldstein RB, Kilpatrick SJ et al: The role of velamentous cord insertion in the etiology of twin-twin transfusion syndrome. Obstet Gynecol 81: 569, 1993 |
|
Qureshi F, Jacques SM: Marked segmental thinning of the umbilical cord vessels. Arch Pathol Lab Med 118: 826, 1994 |
|
Robinson LK, Jones KL, Benirschke K: The nature of structural defects associated with velamentous and marginal insertion of the umbilical cord. Am J Obstet Gynecol 146: 191, 1983 |
|
Minakami H, Akahori A, Sakurai S et al: Umbilical vein thrombosis as a possible cause of perinatal morbidity or mortality: report of two cases. J Obstet Gynaecol Res 27: 97, 2001 |
|
Machin GA, Ackerman J, Gilbert-Barness E: Abnormal umbilical cord coiling is associated with adverse perinatal outcomes. Pediatr Dev Pathol 3: 462, 2000 |
|
Vern TZ, Alles AJ, Kowal-Vern A et al: Frequency of factor V(Leiden) and prothrombin G20210A in placentas and their relationship with placental lesions: Hum Pathol 31:1036, 2000 |
|
Fleming AD, Salafia CM, Vintzileos AM et al: The relationships amongst umbilical arterial velocimetry, fetal biophysical profile and placental inflammation in preterm premature rupture of the membranes. Am J Obstet Gynecol 164: 38, 1991 |
|
Byrne J, Blanc WA: Malformations and chromosome anomalies in spontaneously aborted fetuses with single umbilical artery. Am J Obstet Gynecol 151: 340, 1985 |
|
Heifetz SA: Single umbilical artery: A statistical analysis of 237 autopsy cases and review of the literature. Perspect Pediatr Pathol 8: 345, 1984 |
|
Jauniaux E, DeMunter C, Vanesse M et al: Embryonic remnants of the umbilical cord: Morphologic and clinical aspects. Hum Pathol 20: 458, 1989 |
|
Altshuler G, Hyde S: Meconium-induced vasocontraction: A potential cause of cerebral and other fetal hypoperfusion and of poor pregnancy outcome. J Child Neurol 4: 137, 1989 |
|
Naeye RL: Can meconium in the amniotic fluid injure the fetal brain? Obstet Gynecol 86: 720, 1995 |
|
Altshuler G, Arizawa M, Molnar-Nadasdy G: Meconium-induced umbilical cord vascular necrosis and ulceration: A potential link between the placenta and poor pregnancy outcome. Obstet Gynecol 79: 760, 1992 |
|
Higginbottom MC, Jones KL, Hall BD, Smith DW: The amniotic band disruption complex: Timing of amniotic rupture and variable spectra of consequent defects. J Pediatr 95: 544, 1979 |
|
Kalousek D: Amniotic band syndrome in previable fetuses. Pediatr Pathol 7: 488, 1987 |
|
Heifetz SA: Strangulation of the umbilical cord by amniotic bands: Report of 6 cases and literature review. Pediatr Pathol 2: 285, 1984 |
|
Miller ME, Graham JM, Higginbottom MC, Smith DW: Compression defects from early amnion rupture: evidence for mechanical teratogenesis. J Pediatr 98: 292, 1981 |
|
Seeds JW, Cafelo RC, Herbert WP: Amniotic band syndrome. Am J Obstet Gynecol 144: 243, 1982 |
|
Irving WL, Doublestein GL: Congenital amniotic band syndrome: report of a familial recurrence. J Am Osteopathic Assoc 88: 891, 1988 |
|
Moerman P, Fryns JP, Vandenberghe K, Lauweryns JM: Constrictive amniotic bands, amniotic adhesions, and limb-body wall complex discrete disruption sequences with pathogenetic overlap. Am J Med Genet 42: 470, 1992 |
|
Colpaert C, Bogers J, Hertveldt K et al: Limb-body wall complex: 4 new cases illustrating the importance of examining placenta and umbilical cord. Pathol Res Pract 196: 783, 2000 |
|
Martinez-Frias ML, Bermejo E, Rodriguez-Pinilla E, Frias JL: Maternal and fetal factors related to abnormal amniotic fluid. J Perinatol 19: 514, 1999 |
|
Mashiah N, Levit A, Sherer DM et al: Two rare complications of simultaneously occurring placenta previa and placenta percreta. Acta Obstet Gynecol Scand 67: 655, 1988 |
|
Salafia CM, Sherer DM, Minior VK et al: Histologic evidence of abnormally deep trophoblast invasion: Clinical correlations. Obstet Gynecol 87: 444, 1996 |
|
Jacques SM, Qureshi F, Trent VS, Ramirez NC: Placenta accreta: Mild cases diagnosed by placental examination. Int J Gynecol Pathol 15: 28, 1996 |
|
Fox H: The villous cytotrophoblast as an index of placental ischaemia. J Obstet Gynecol Br Commonw 71: 885, 1967 |
|
Tominaga T, Page EW: Accomodation of the human placenta to hypoxia. Am J Obstet Gynecol 94: 679, 1966 |
|
Zhou Y, Chiu K, Brescia RJ et al: Increased depth of trophoblast invasion after chronic constriction of the lower aorta in Rhesus monkeys. Am J Obstet Gynecol 169: 224, 1993 |
|
Wenstrom KD, Gall SA: Incidence, morbidity and mortalilty, and diagnosis of twin gestations Clin Perinatol 15: 1, 1988 |
|
Bronsteen R, Goyert G, Bottoms S: Classification of twins and neonatal morbidity. Obstet Gynecol 74: 98, 1989 |
|
Johnson SF, Driscoll SG: Twin placentation and its complications. Sem Perinatol 10: 9, 1986 |
|
McCulloch K: Neonatal problems in twins. Clin Perinatol 15: 141, 1988 |
|
Newton ER: Antepartum care in multiple gestations. Sem Perinatol 10: 19, 1986 |
|
Kleinman JC, Fowler MG, Kessel SS: Comparison of infant mortality among twins and singletons: United States 1960 and 1983. Am J Epidemiol 133: 133, 1991 |
|
Benirschke K, Kim CK: Multiple pregnancy (first of two parts). N Engl J Med 188: 1276, 1973 |
|
Baldwin VJ: Pathology of multiple gestation. In Wigglesworth J, Singer DB (eds): Textbook of Fetal and Perinatal Pathology. Cambridge, Blackwell Scientific Publications, 1991 |
|
Chen SE, Trupin L, Trupin S: Antepartum rupture of diamniotic membranes separates monozygotic twins: A case report. J Reprod Med 39: 67, 1994 |
|
Pinar H, Sung CJ, Oyer CE, Singer DB: Reference values for singleton and twin placental weights. Pediatr Pathol Lab Med 16: 901, 1996 |
|
Soernes T, Bakke T: The length of the human umbilical cord in twin pregnancies. Am J Obstet Gynecol 157: 1229, 1987 |
|
Robertson EG, Neer KJ: Placental injection studies in twin gestation. Am J Obstet Gynecol 147: 170, 1983 |
|
Landy HJ, Weiner S, Corson SL et al: The “vanishing twin”: Ultrasonographic assessment of fetal disappearance in the first trimester. Am J Obstet Gynecol 147: 170, 1983 |
|
Dudley DKL, D'Alton ME: Single fetal death in twin gestation. Semin Perinatol 10: 65, 1986 |
|
Cherouny PH, Hoskins IA, Johnson TRB et al: Multiple pregnancy with late death of one fetus. Obstet Gynecol 74: 318, 1989 |
|
Patten RM, Mack LA, Nyberg DA et al: Twin embolization syndrome: Prenatal sonographic detection and significance, Radiology 173: 685, 1989 |
|
Benirschke K: Intrauterine death of a twin: Mechanisms, implications for surviving twin, and placental pathology. Semin Diagn Pathol 10: 222, 1993 |
|
Grafe MR: Antenatal cerebral necrosis in monochorionic twins. Pediatr Pathol 13: 15, 1993 |
|
Enbom JA: Twin pregnancy with intrauterine death of one twin. Am J Obstet Gynecol 131: 267, 1978 |
|
Lembet A, Selam B, Gaddipati S et al: Shortened gestational age following multifetal pregnancy reduction: Can chronic placental inflammation be the explanation? J Matern Fetal Med 10: 149, 2001 |
|
Gardner MO, Goldenberg RL, Cliver SP et al: The origin and outcome of preterm twin pregnancies. Obstet Gynecol 85: 553, 1995 |
|
Naeye RL, Benirschke K, Hagstrom JWC et al: Intrauterine growth in twins as estimated from liveborn birth weight data. Pediatrics 37: 409, 1966 |
|
Eberle AM, Levesque D, Vintzileos AM et al: Placental pathology in discordant twins. Am J Obstet Gynecol 169: 931, 1993 |
|
Jacques SM, Qureshi F: Chronic villitis of unknown etiology in twin gestations. Pediatr Pathol 14: 575, 1994 |
|
Benirschke K, Kim CK: Multiple pregnancy (second of two parts). N Engl J Med 188: 1329, 1979 |
|
Little J, Bryan E: Congenital anomalies in twins. Semin Perinatol 10: 50, 1986 |
|
Heifetz SA: The placenta. In Stocker JT, Dehner LP (eds): Pediatric Pathology. Philadelphia, JB Lippincott, 1992 |
|
Lage JM, Vanmarter LJ, Mikhail E: Vascular anastomoses in fused, dichorionic twin placentas resulting in twin transfusion syndrome. Placenta 10: 55, 1989 |
|
Blickstein I: The twin-twin transfusion syndrome. Obstet Gynecol 76: 714, 1990 |
|
Genest DR, Lage JM: Absence of normal-appearing proximal tubules in the fetal and neonatal kidney: Prevalence and significance. Hum Pathol 22: 147, 1991 |
|
Zosmer N, Bajoria R, Weiner E et al: Clinical and echographic features of in utero cardiac dysfunction in the recipient twin in twin-twin transfusion. Br Heart J 72: 74, 1994 |
|
Sala MA, Matheus M: Placental characteristics in twin transfusion syndrome. Arch Gynecol Obstet 246: 51, 1989 |
|
Bendon RH, Siddiqi T: Clinical pathology conference: Acute twin-to-twin in utero transfusion. Pediatr Pathol 9: 591, 1991 |
|
Danskin WH, Neilson JP: Twin-to-twin transfusion syndrome: What are appropriate diagnostic criteria? Am J Obstet Gynecol 161: 365, 1989 |
|
Bruner JP, Rosemond RL: Twin-to-twin transfusion syndrome: A subset of the twin oligohydtramnios/polyhydramnios sequence (TOPS). Am J Obstet Gynecol 169: 925, 1993 |
|
Bendon RW: Twin transfusion: Pathological studies of the monochorionic twin pairs. Pediatr Pathol Lab Med 15: 363, 1995 |
|
Baldwin VJ: The pathology of monochorionic monozygosity. In Pathology of Multiple Pregnancy, Chapter 8. New York, Springer Verlag, 1994 |
|
Bajoria R, Wigglesworth J, Fisk NM: Angioarchitecture of monochorionic placentas in relation to the twin-twin transfusion syndrome. Am J Obstet Gynecol 172: 856, 1995 |
|
Van Allen MI, Smith DW, Shepard TH: Twin reversed arterial perfusion (TRAP) sequence: A study of 14 twin pregnancies with acardius. Semin Perinatol 7: 285, 1983 |
|
Popek EJ, Strain JD, Neumann A, Wilson H: In utero development of pulmonary artery calcification in monochorionic twins: A report of three cases and discussion of the possible etiology. Pediatr Pathol 13: 597, 1993 |
|
Moore TR, Sale SA, Benirschke K: Perinatal analysis of forty-nine pregnancies complicated by acardiac twinning. Am J Obstet Gynecol 163: 907, 1990 |
|
Wolf HK, MacDonald J, Bradford WB et al: Acardius anceps with evidence of intrauterine vascular occlusion: Report of a case and discussion of the pathogenesis. Pediatr Pathol 11: 143, 1991 |
|
Stephens TD, Spall R, Urfer AG et al: Fetus amorphus or placental teratoma. Teratology 40: 1, 1989 |
|
Kiely JL, Kleinman JC, Kiely M: Triplets and higher-order multiple births, time, trends, and infant mortality. Am J Dis Child 146: 862, 1992 |
|
Pinar H, Stephens M, Sung CJ et al: Reference values for triplet placental weights. Pediatr Dev Pathol 4: 417A, 2001 |