Obstetric and Fetal Pharmacology
Authors
INTRODUCTION
One of the most neglected areas in drug development and clinical pharmacology involves the study of drugs given to pregnant women.1 Consequently, only a handful of drugs have been approved by the Food and Drug Administration (FDA) for use in pregnancy, or contain obstetric dosing information in the product or package insert with the possible exception of adverse fetal effects.2 Sadly, because women are not studied, dosing recommendations are frequently incorrect.3
Pregnancy poses a unique situation because drugs are generally given to treat the mother, however, the fetus is always a recipient and may be the therapeutic target4 as in AIDS.5 Concern over the fetal consequences of maternal drug therapy has totally overshadowed the need to study the biodisposition of drugs in the mother.6 Conceptually, the mother has been viewed as a woman that “carries” the fetus and not as a woman whose pregnancy may significantly alter the biodisposition of drugs. The pharmacologic and toxic effects of drugs on the mother, placenta, and the fetus are governed by a complex but integrated set of variables consisting of mother, uterus, placenta, amniotic fluid, and fetus.7 Because all components of this unit are constantly changing throughout pregnancy, the system truly presents a formidable pharmacologic challenge.8
This chapter summarizes the clinical implications of obstetric and fetal pharmacology, and concentrates on relating the pharmacodynamics and pharmacokinetics of drugs to the physiologic changes and pathophysiologic disturbances that occur during pregnancy.9 Concepts are emphasized to enable the obstetrician and perinatologist to provide optimal therapeutics without adverse effects based on evidenced-based pharmacology.
GENERAL PRINCIPLES
Drugs undergo a series of interactions in the body before combining with specific tissue receptors and producing the desired pharmacologic effect. A number of variables can modify the intensity and duration of pharmacologic effect: rate and extent of absorption, volume of distribution, rate and nature of metabolism and excretion, and interaction with other compounds.
Factors that determine the rate and the percentage of the compound that is absorbed (bioavailability) are the physiochemical characteristics of the drug, its rate of dissolution, the gastric and intestinal pH, gastric emptying time, composition of intestinal contents, intestinal motility, and mesenteric blood flow.10 Determinant factors of absorption by other routes (i.e., intramuscular, subcutaneous, epidural) are degree of ionization, physiochemical composition, water or lipid solubility, and blood flow at the site of injection. After absorption, drugs enter the intravascular system and either circulate in free form or are bound to plasma proteins to differing degrees depending on their binding characteristics and other competing ligands.
Distribution of unbound drugs throughout the body is frequently a rapid process that allows diffusion equilibrium to be quickly established between blood and other body compartments. In some situations, however, the access of a drug to the sites of its pharmacologic action may require considerable time. Under these circumstances, measurements of drug concentrations in blood may not correlate with pharmacologic effects (at least not until “steady state” has been reached).
Among other factors, drug distribution is influenced by lipid solubility, degree of ionization, blood flow, and binding affinities to proteins in plasma and specific tissues.11 From the pharmacologic standpoint, a drug is eliminated either by excretion or by metabolic biodegradation into pharmacologically inactive metabolites. Although renal excretion of unchanged drugs is by far the most important excretory route, there are several other excretory pathways (such as biliary excretion or alveolar elimination) used by certain compounds. These excretory pathways may assume greater importance in certain pathologic conditions that preclude the use of the primary excretory route.
PHARMACOLOGY OF PREGNANCY
Normal human pregnancy is accompanied by such remarkable physiologic changes that drug disposition and effect may be entirely different from those in nonpregnant patients. These differences are important not only for maternal therapy but also for understanding the effects of fetal drug exposure, and potential fetal therapeutics.
Role of gender
The effects of the pregnant state on the disposition and action of drugs are superimposed on the changes associated with the female sex. Sex differences regarding drug disposition in experimental animals have been known for more than 60 years, but it was not until 1993 that the FDA encouraged the inclusion of women in clinical trials.12 There are striking differences in body processes between men and women. Physiologic differences between the sexes may explain variations in the absorption of drugs. Compared to men, women have slower gastric emptying time and prolonged colonic transit time. These differences may be heightened during pregnancy. There are also differences in drug biotransformation.
A multienzyme system is responsible for the degradation of hydrophobic molecules. In a sequential manner, hydrophobic molecules are biotransformed by phase I enzymes and then conjugated by phase II enzymes to produce water-soluble products. The cytochrome P450 superfamily (or CYP) is the major phase I group of isoenzymes. These enzymes are expressed mostly in the liver but also to a lesser extent in other tissues (e.g., intestine). The expression pattern of different CYP isoforms differs between the sexes. For example, the cytochrome P450 CYP3A4 is more active in women than in men.13 Theophylline and acetaminophen, which are metabolized by CYP3A4, are eliminated faster by women. Other drugs, such as diazepam, caffeine, and some anticonvulsants, metabolized by CYP2C19 or CYP1A2 appear to be metabolized faster in men than in women.14 Gender differences in drug biodisposition have been linked to variations in sex hormones. Sex differences in the receptors and transporters have not been systematically studied. Studies in animal models have shown sex differences but the results have not been validated in human studies.
There are also sex differences in the sensitivity to drugs.15 Opioids such as pentazocine show a greater drug response in women, whereas ibuprofen produces a better response in men.16, 17 In addition, there are gender differences in the incidence of adverse drug reactions. For example, drug-induced torsades des pointes and the cough induced by angiotensin-converting enzyme inhibitors occur more commonly in women.18, 19 A distinction between pharmacokinetics, pharmacodynamics, and sex-dependent differences is not always possible. Similarly the influence of genetics remains to be determined.
Mother-fetus: a two-compartment system
A fundamental aspect of fetal pharmacology is that of fetal dose. The amount and rate of transfer of drugs to the fetus determine the presence or absence of pharmacologic or toxic effects.
With the rare exception of drugs injected directly into the fetal compartment, the path a drug must take from its administration to the mother is across the maternal organism to its site of action in the fetus.
This multicompartment system is especially complicated because it does not represent a constant relationship but one that is continuously changing throughout pregnancy (Fig. 1).
|
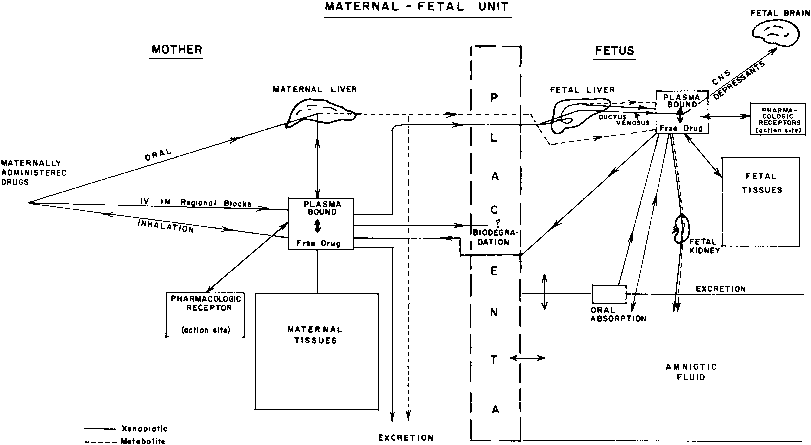
THE MOTHER
Absorption
The gastrointestinal absorption of drugs has not been systematically studied during pregnancy. Reports in the literature suggest that a generalized malabsorption state may be induced or exacerbated during pregnancy.20, 21 Both gastric emptying time and gastrointestinal transit times are prolonged, probably due to the high levels of progesterone. There is also indirect evidence that absorption of certain compounds such as digitoxin, salicylamide, and phenytoin may be delayed in pregnant patients.22, 23, 24
The confluence of different physiologic alterations during pregnancy may potentiate the effect of individual changes. For example, the increased residence time due to the decrease in intestinal motility could lead to a decreased bioavailability because of an increase in gut metabolism. The latter may occur if the activity of gut CYP3A4 mirrors the increase in the activity of hepatic CYP3A4 that has been documented during pregnancy.
The complexities of performing bioavailability studies in pregnancy can be simplified with the use of stable isotopes. The intravenous injection of a stable isotope-labeled drug coupled with the concomitant oral administration of unlabeled drug permits the simultaneous determination of both drug profiles and therefore minimizes the variability associated with two separate studies.25 The normal circulatory adjustments that occur during pregnancy are therefore likely to influence the extent of drug absorption. These adjustments can be expected to be more influential toward the end of pregnancy. Indeed, the absorption rate of meperidine after intramuscular administration has been found to be slower in women during labor than in nonpregnant controls.26, 27 On the other hand, circulatory changes are on occasion pharmacologically advantageous. Studies have shown that pregnant women at term require 40–50% less drugs for peridural block because at term the capacity of the peridural space is reduced by the physiologic engorgement of the internal vertebral plexus.
Distribution
The remarkable changes in the volume of water and composition of body compartments, coupled with the hemodynamic adjustments that occur during pregnancy, set a background for drug distribution that is quite different from that present in nonpregnant persons. Dilutional hypoalbuminemia, especially in the last trimester, is mainly responsible for a decrease in drug-binding capacity and a consequent increase in body distribution.28 As a corollary, the acceptable therapeutic range of total plasma concentration of drugs exhibiting decreased protein binding (i.e., phenytoin) is lower than in the nonpregnant state. The great interindividual variability in the distribution of drugs such as meperidine given during labor may be attributed to variations in the hemodynamic makeup of different women.29
Metabolism
The rate and extent of drug biotransformation depend on hepatic blood flow, activities of drug-metabolizing enzymes systems, and hormonal influence. Hepatic blood flow measured in absolute terms does not appear to be altered during pregnancy. Proportionally, however, the percentage of cardiac blood flow reaching the liver is decreased.30
Knowledge of drug metabolism during pregnancy and in women in labor is scanty and far from satisfactory. The sex hormone changes characteristic of pregnancy and placental hormones influence hepatic drug-metabolizing enzymes' activities during gestation. Clinical studies using metabolic probes have shown that CYP1A2, xanthine oxidase, and N-acetyltransferase activities are decreased, but CYP3A4 is increased during pregnancy.31 N-acetyltransferase activity, measured using caffeine as a probe, has been found to be reduced by about 30% in late pregnancy compared to the enzyme activity during the puerperium.31
The metabolism of drugs catalyzed by select cytochrome P450 (CYP) isoenzymes (i.e., CYP3A4, CYP2D6, and CYP2C9) and uridine diphosphate glucuronosyltransferase (UGT) isoenzymes (i.e,. UGT1A4 and UGT2B7) is increased during pregnancy. Dosages of drugs predominantly metabolized by these isoenzymes may need to be increased during pregnancy in order to avoid loss of efficacy. In contrast, CYP1A2 and CYP2C19 activity is decreased during pregnancy, suggesting that dosage reductions may be needed to minimize potential toxicity of their substrates.32, 33 There are limitations to the available data. Current knowledge is based primarily on observational studies, many including small numbers of women. For some isoenzymes, the effect of pregnancy on only one drug has been evaluated. The full-time course of pharmacokinetic changes during pregnancy is often not studied sequentially during pregnancy. Drugs eliminated by non-CYP or non-UGT pathways or multiple pathways will need to be evaluated individually.34
A differential effect on the activity of CYP2D6 was reported in pregnant subjects. In both homozygous and heterozygous fast metabolizers, CYP2D6 activity increased, whereas the activity of this polygenic enzyme decreased in homozygous poor metabolizers.35 Demethylation of meperidine occurs less readily in women in labor.36 Conversely, other metabolic pathways may be inducible. The reported increase in plasma clearance of phenytoin and phenobarbital during pregnancy has been attributed to an increase in metabolic rate.37 The increased microsomal oxidation of carbamazepine and phenytoin during pregnancy is not associated with a proportional increase in the subsequent hydroxylation and glucuronidation of the initial metabolite.38
Glucuronide conjugation of endogenous substances and xenobiotics (compounds foreign to the body) is inhibited during late pregnancy.39, 40 The inhibition of glucuronosyltransferase activity may be related to the high tissue levels of progesterone and pregnanediol. Glucuronidation of salicylamide has been found to be depressed in parturients. There is indirect evidence, however, of increased glucuronidation of other substrates, such as zidovudine.41 The increased clearance of labetalol during pregnancy has been linked to an increase in labetalol glucuronidation. UGT1A1 and UGT2B7 have been identified as the enzymes producing labetalol glucuronides possibly under the modulating influence of progesterone.42
Excretion
Although almost all maternal physiologic systems undergo adjustments during pregnancy, perhaps the greatest change is seen in the renal system. The profound changes in function are likely to affect the renal excretion of drugs. The increase in glomerular filtration observed during pregnancy is counterbalanced to a significant extent by modifications in tubular reabsorptive capacity. Of greatest importance from the pharmacokinetic standpoint are diurnal variations in function.
These factors are likely to markedly influence the “dosing” of drugs transferred to the fetus. Although for the purpose of this discussion, absorption, distribution, and elimination of drugs in the maternal organism have been considered separately, the interplay among these factors determines the time course of maternal plasma levels. Little is known about the extent to which complications of pregnancy modify drug disposition in the mother.
ROLE OF THE PLACENTA
The placenta has been regarded as the somewhat passive and inert barrier to the transfer of drugs between the fetal and maternal compartments. However, recent work has uncovered a bewildering number of complex functions affecting both maternal and fetal physiology.43, 44 Two aspects of placental pharmacology assume equal importance: the transfer and disposition of xenobiotics reaching the organ from the maternal and fetal side, and the biodegradation properties affecting xenobiotics or being affected by them. The three major factors affecting drug transfer across the placenta are physiochemical characteristics of the compound, pharmacologic properties of the placental tissue, and maternal and fetal placental blood flow.
Physiochemical characteristics include molecular weight, lipid solubility, degree of ionization, molecular configuration, and tissue protein properties. Generally, lipophilic substances and compounds with low molecular weight tend to diffuse rapidly into the fetal circulation. Poorly ionized drugs at physiologic pH, such as thiopental, reach the fetal circulation quite rapidly. Certain compounds, such as the sympathomimetic agents, salbutamol, ritodrine, and norepinephrine, appear to have a low transfer rate despite their small molecular weights (170–290). Still, sufficient amounts of both salbutamol and ritodrine are transferred to produce fetal tachycardia.45, 46
The limited transfer rate of norepinephrine may be attributed in part to biodegradation by placental tissue. The purported placental impermeability to polar compounds is relative rather than absolute. The high lipid solubility of certain compounds, such as salicylates, allows for rapid transfer despite being almost 100% ionized at physiologic pH. Xenobiotics cross the placenta by different transfer mechanisms: simple diffusion, facilitated diffusion, active transport, and pinocytosis. The metabolic conversion by the placenta of one compound into another compound that in turn may be transferred cannot be discounted. Most drugs cross the placenta by simple diffusion at a rate that is directly related to the difference between the maternal and fetal blood concentrations. Recent studies have shown that the syncytiotrophoblast expresses membrane proteins that act as drug transporters.47 P-glycoprotein, multidrug resistant proteins (MRP 1–3), and breast cancer resistant protein (BPCRP) may be involved in the efflux of drugs into the maternal circulation. P-glycoprotein, an ATP-dependent drug efflux pump, is present in the brush border of the syncytiotrophoblast.48 Drug transport by P-glycoprotein is unidirectional from the fetal to the maternal side and thus protects the fetus from toxic compounds. A wide variety of drugs are substrates for this transporter (e.g., digoxin, verapamil, chemotherapeutic agents). Transporters in the opposite direction have not been sufficiently characterized.
Transporters, including organic anion transporter (OATP) serotonin transporter, norepinephrine transporter (NET), and several organic transporters are also expressed in the placenta, but their pharmacologic role remains unknown. The role of the carnitine transporter is in the delivery of carnitine to the fetus, however, a number of pharmacologic active compounds such as verapamil and cephaloridine may utilize this transporter. Bile acid transporters are present in the placenta and may be important in the efflux of compounds back into the maternal circulation. The physiologic role of placental nucleoside transporters (ENT 1 and 2) is to facilitate the transport of purine and pyrimidine nucleosides from the mother to the fetus. A significant number of drugs including anticancer drugs may utilize these transport systems. There are transport systems in animal studies for which no endogenous susbstrates have been identified.49, 50, 51, 52
The placenta undergoes continuous structural changes during its life span that are likely to significantly affect rates of drug diffusion. Studies in the pregnant rodent seem to indicate that drug transfer is lowest in midgestation and peaks at the beginning and end of pregnancy.53 There is little information on the effect of placental aging on drug transfer in normal human pregnancy, let alone those changes that occur during abnormal conditions, when drugs are most often prescribed. The relative maternal and fetal blood flow through the placenta is of paramount importance in determining the rate of drug transfer from mother to fetus and vice versa.
Adequate measurements of uterine blood flow are flawed by technical difficulties. Despite this, several studies have shown an increase in uterine flow per kilogram of uterine weight toward term. When data are analyzed in terms of uterine blood flow per kilogram of fetus, however, a decrease is demonstrated at term. The typical time course of uterine and fetal plasma concentrations usually follows the pattern:
- Establishment of a maternal–fetal concentration gradient;
- Equilibration phase, in which the highest fetal drug concentration will depend on the placental factors discussed above;
- Fetal drug elimination phase. During this period, the combined effects of maternal drug biodegradation and elimination lower maternal drug concentrations, creating a fetal–maternal gradient and reversing the direction of drug transfer across the placenta.
Delivery can occur at any point during this sequential pattern, and its timing will determine the amount of drug present and the ability of the newborn to handle xenobiotics. Many factors can influence maternal and fetal hemodynamics, thereby disturbing maternal and fetal drug distribution. Those affecting maternal hemodynamics are briefly reviewed here. A decrease in uteroplacental blood flow may be secondary to vasoconstriction of myometrial arterioles or obstruction of uterine venous outflow. The amount of drug transfer to the fetus, especially after a single intravenous pulse injection, will vary depending on the type of blood flow obstruction and the temporal relationship between drug administration and the onset of uterine hypoperfusion. For drugs given before the onset of uterine blood flow obstruction, myometrial arteriole vasoconstriction will tend to protect the fetus, whereas venous obstruction, by allowing a longer period of placental residency time, will result in increased fetal drug extraction. Alterations in uterine blood flow of particular interest are those related to abnormal labor, excessive uterine activity (spontaneous or oxytocin-induced), vasoactive drugs, or vena cava compression and supine hypotension, as may occur at the time of removal of amniotic fluid.
Pathophysiologic conditions, such as preeclampsia, hypertension, and diabetes, which may be associated with impaired uteroplacental blood flow, can be expected to decrease drug transfer across the placenta. On the other hand, these pathophysiologic conditions often are associated with profound fetal hemodynamic changes that favor drug distribution to the fetal brain.
The demonstration that the human placenta is capable of metabolizing xenobiotics spurred a burst of investigative activity. Placental CYP1A1 is inducible by maternal smoking. CYP4B1 and CYP19 may contribute to the metabolism of some drugs.54 The picture emerging from the available research, however, indicates that although the placenta is a major metabolic organ for the biotransformation of endogenous substances, especially steroidal hormones, its contribution to the overall degradation of drugs during pregnancy is quantitatively meager.55 The demonstration that foreign organic substances could undergo oxygenation in human placental tissues raises the possibility that xenobiotics and endogenous steroids might share common biotransformation reactions. The balance of present evidence, however, refutes this contention and supports the existence of separate P450 species of isoenzymes for the catalysis of xenobiotic and steroidal hydroxylation reactions. The discovery that the placental mono-oxygenase activities are inducible by maternal cigarette smoking and not by other inducers is of considerable interest in this regard.56 The placental tissue of smokers contains bioactivating enzymes that catalyze the formation of metabolites that covalently bind to DNA57 or produce mutations in Salmonella typhimurium.58 It remains a challenge for researchers to determine whether the demonstrated bioactivating capacity of the placenta allows the formation of reactive metabolites of chemical carcinogens and mutagens that could damage the embryo or the fetus.
THE FETUS
The study of the pharmacologic and toxic effects of drugs on the human fetus has been limited by lack of accessibility and by societal and ethical constraints against human fetal research. Imaging technology has been applied to diagnose fetal disorders59, 60 and may be used to monitor fetal therapeutic interventions. The effects of drugs given during the first trimester are not considered in this review.
During its intrauterine existence, the fetus may be exposed to a multitude of chemicals, including drugs, herbal medicines, alcohol, caffeine, tobacco, and environmental pollutants. The available data have been gathered by different approaches: (1) in vitro studies of drug metabolism using subcellular fractions (e.g., liver, adrenal glands) or isolated hepatocytes from aborted fetuses; experiments with isolated cells yield more meaningful information than those performed with subcellular fraction of tissues because the enzyme system operates under more physiologic conditions; (2) studies of drug disposition or distribution in fetuses with lethal malformations (e.g., anencephaly) or stillbirths; and (3) postnatal pharmacokinetic studies of drugs transferred in utero before birth. Postnatal in vivo studies using plasma elimination curves are useful for analysis of drug distribution and effects of different dose-delivery intervals. They are, however, of limited value for studies of drug metabolism because metabolites may be present due to transfer across the placenta (see Fig. 1), and there is a marked shift in the balance between metabolic and excretory pathways of elimination immediately after delivery.
The late gestational period, of particular interest for this review, is also the time of fetal life that is largely beyond the reach of investigation. The study of preterm stillborn infants is of limited value because of the pathologic circumstances under which these studies are performed. Pregnant sheep and primate models have significantly contributed to the present understanding of fetal drug disposition.
Distribution
The fetal circulation is so unusual that it greatly modifies drug distribution. Xenobiotics enter the fetus mostly through the umbilical vein. Most (60–80%) of the umbilical vein flow perfuses the liver; the remainder is shunted to the inferior vena cava by way of the ductus venosus. Dilution of umbilical venous blood in the right atrium and shunting across the foramen ovale and ductus arteriosus into the systemic circulation significantly affect fetal drug distribution. Fetal hepatic blood flow is quite variable and can be markedly reduced by hypoxia. Under these circumstances, drugs bypass the liver and can reach high concentrations in the fetal brain and other tissues. The physiologic characteristics of the fetal circulation are such that even under normal circumstances, the brain of the fetus receives a larger share of the cardiac output than the brain at any other period in life. Redistribution of the cardiac output away from nonessential vascular beds during asphyxia may have a profound effect on drug distribution to the fetal brain and heart. The recent discovery of the lack of autoregulation of the cerebral circulation in preterm infants may render this organ even more vulnerable to large infusions of drugs under hypoxic conditions.
The composition of fetal blood itself may produce changes in drug distribution that are clinically significant. Fetal serum proteins usually bind drugs to a lesser degree than adult proteins.61, 62 This results in an increase in the proportion of unbound free drug that is responsible for pharmacologic effect. This difference in binding affinity is not universal. Neonatal and adult sera bind digoxin to a similar extent, and fetal red cells take up more trichloroethylene than maternal erythrocytes. The reduced plasma protein binding of various drugs is due to a combination of factors: reduced total plasma protein, persistence of fetal albumin with lower binding affinity for drugs, lower concentration of immunoglobulins, and competitive binding of endogenous substances and compounds of maternal origin.
The kinetics of drug distribution to various tissues and body compartments is profoundly affected by the remarkable changes in fetal body composition that occur during pregnancy. Total body water content decreases from about 94% of total body weight at 16 weeks of gestation to about 76% at term. A progressive decrease in extracellular water is responsible for the change. Fat, which is virtually absent in fetuses weighing less than 1000 g, is accrued during the last trimester. Even at term, fat tissue is relatively scarce (15% of body weight) and contains more water than similar tissue from older individuals. The relative paucity of fat tissue limits the distribution in fetal tissues of lipid-soluble compounds such as barbiturates, general anesthetics, and diazepam. The changes in the composition of individual organs and their relative contribution to the overall body composition further complicate the understanding of drug distribution in the fetus. In relation to total body mass, the newborn has less skeletal muscle and greater brain and liver tissue than the adult. The composition of fetal brain tissue is also quite different from adults in that its myelin content is low and its water content is relatively large. These compositional changes, coupled with high cerebral blood flow with preferential perfusion of brain stem structures, are likely to lead to different distribution of flow-dependent lipophilic compounds in the fetal brain.
Amniotic fluid dynamics
Fetal drug clearance depends on the combined contribution of fetal renal excretion and hepatic metabolism. The interposition of the amniotic fluid compartment between mother and fetus adds an element of complexity to the distribution and disposition of drugs reaching the fetus.
Although the possibility of direct maternal amniotic fluid exchange cannot be excluded, if it occurs it is likely to be small. Most exchange occurs through the fetus. In this, the amniotic fluid compartment should be considered as one of the fetal excretory pathways, but because of fetal swallowing, recirculation is likely to occur. Sequestration of drugs in the fetal gut must also be considered. Recent interest in fetal therapeutics has raised the possibility of amniotic fluid infusion of drugs. Although promising, studies are still preliminary and in the animal testing stage.63 The limited data available suggest that water-soluble compounds enter the fetal compartment slowly, probably by fetal swallowing. In contrast, lipid-soluble compounds introduced into the amniotic sac reach the fetus much more rapidly and in significant amounts. It has been speculated that lipid-soluble molecules enter the fetus through the skin or the fetal surface of the placenta, or both. Systematic studies are needed to clarify the various factors determining drug absorption by this route.
Metabolism of xenobiotics
Research has clearly demonstrated that the human fetus is endowed with a well-developed system of xenobiotic metabolizing enzymes.64 Enzymatic activities, however, are significantly reduced in comparison with adult values but appear to increase with advancing fetal and postnatal age. The most significant pathways of drug biotransformation are oxidative reactions. The enzymatic systems that are located in the microsomal fraction of hepatocytes also catalyze the biotransformation of fatty acids, bile acids, and steroid hormones. It is likely that endogenous substances have a much higher affinity for the terminal oxidase in the microsomal system than do xenobiotics. The different components of the microsomal oxidizing system that function as an electron transport chain are present in the human fetal liver at levels one-fifth to four-fifths those of the adult. Cytochrome P450, the last component in the chain, is already present in the human fetal liver by the latter part of the first trimester.65 Studies of postmortem liver samples have shown the total P450 content remains stable from the first trimester of pregnancy in the fetus until the first year of postnatal life.66
Studies comparing cytochrome P450-dependent enzyme activities of adult liver, placenta microsomes, and fetal liver suggest that the differences between these tissues are due to the existence of different tissue-specific isoenzymes.67 CYP2C proteins are absent from the fetal liver. Hydroxylation of tolbutamide and demethylation of diazepam depend on CYP2C activity. The P450 subfamily CYP3A includes three isoforms: CYP3A4, CYP3A5, and CYP3A7. CYP3A7 is mostly expressed in the fetal liver and is replaced at birth by CYP3A4.68 CYP1A2 and CYP2D6 are not expressed in the fetus. The N-demethylation of caffeine and theophylline is particularly deficient, probably because CYP1A2 is involved in their metabolism.69
Knowledge of the fetal ontogenic profiles of CYP proteins is an important step toward the estimation of risk associated with fetal drug exposure. Substrates that have been metabolized by phase I (e.g., CYP proteins) enzymes are further biotransformed by phase II conjugation drug-metabolizing enzymes. Among synthetic phase II reactions, sulfate conjugation and glycine conjugation are especially efficient and approach rates found in adults. It has been suggested that these well-developed reactions compensate for the deficiency in glucuronic acid conjugation by glucuronyl transferases (UGTs) in fetal life. The large amounts of sulfate conjugates of steroids in human fetal tissues support this hypothesis. UGTs consist of at least 18 different isoforms. UGT1A1 is responsible for the conjugation of bilirubin, steroids, and several drugs. UTG2B7 is the most important isoform responsible for the metabolism of morphine, opioid derivatives, lorazepam, and nonsteroidal anti-inflammatory agents.70 Although UGTs enhance renal excretion of hydrophilic intermediates, glucuronide metabolites may be potentially toxic. For example, glucuronide metabolites of morphine are active (morphine-6-glucuronide is 100 times more potent than morphine and morphine-3-glucuronide is neuroexcitatory). It has been shown that fetal baboons conjugate morphine at both 3-OH and 6-OH positions.71 The formation of zidovudine-glucuronide during fetal infusions in baboons also gives credence to a significant contribution of fetal glucuronidation to the nonplacental clearance of both drugs. Biochemical assays of UGTs in fetal hepatic tissues have shown limited activity (less than 20% of adult activity).72 Studies are needed to compare in vitro UGT activities with rates of metabolite formation in fetal animals. Further, because UGTs are inducible, patterns of their fetal expression may vary during pregnancy.
Molecular and genetic tools are now available to probe for specific CYPs and UGTs and to determine factors responsible for their expression in the fetus. A major goal for future research is the quantification of the nonplacental clearance attributable to fetal metabolism. The possibility of increased concentration of drugs or their metabolites that may even exceed maternal concentrations needs to be considered, particularly when placental transfer back to the mother is compromised. Fetal consequences may ensue if toxic active metabolites are increased.73 Alternatively, fetal drug concentrations may be decreased by fetal metabolism. This may be of significant concern if the fetus is the target of therapy, as in the prevention of HIV perinatal transmission (e.g., zidovudine therapy).
The low levels of blood esterase activities in preterm infants partially explain the cardiac and respiratory depression observed at birth when local anesthetics containing ester bonds are used during labor and delivery.
Extrahepatic sites for xenobiotic metabolism have attracted considerable interest. Present knowledge is preliminary and allows only tentative conclusions. Considerable activity toward some substrates has been found in the adrenal, pancreas, and gonad tissue of the human fetus. Activation of some compounds such as diethylstilbestrol and aromatic hydrocarbons has been related to these findings.74 Inducibility of fetal biotransformation reactions remains a fascinating but modestly understood subject. Xenobiotics, including alcohol administered in large doses and for prolonged periods during the first half of pregnancy, may induce drug-metabolizing enzymes in human fetal liver.75 Pharmacokinetic studies of diazepam and phenytoin in newborns exposed in utero support the concept of transplacental induction by anticonvulsants.76, 77
Excretion
Although the placenta is a major excretory organ, secondary excretory routes may affect the residency time of drugs in the fetal compartment. Xenobiotics or their metabolites may be excreted by the fetal kidneys. Fetal urination contributes significantly to amniotic fluid formation. Hypotonic urine has been obtained from the fetal bladder as early as 12 weeks of gestation. It has been estimated that close to term, the fetus produces 600–800 mL/day of very hypotonic urine (80–140 mOsm/kg water). Water-soluble lipid-insoluble drugs such as antibiotics may be excreted in significant amounts in the fetal urine during the last trimester. The high rate of urine production and high fetal urine concentration of these drugs account for peak amniotic fluid concentrations that are higher than maternal or fetal plasma. A trapping effect of these compounds in the amniotic fluid after excretion by the fetal kidney occurs because equilibration with either fetal or maternal compartments is likely to be a very slow process. Indeed, there are many examples of compounds that accumulate in the amniotic fluid by this mechanism. Ampicillin, penicillin, kanamycin, gentamicin, sulfonamides, methicillin, and some of the cephalosporins behave in this manner.
Evidence supporting this mechanism comes from acute experiments in late pregnancy. When ampicillin was given by pulse intravenous injection to the mother, peak levels in fetal serum were demonstrated 30–60 minutes after injection, and prolonged peak concentrations were found in the amniotic fluid 6–12 hours later. When the fetus was dead, however, only insignificant amounts of ampicillin could be demonstrated in the amniotic fluid.78 The fetal skin may play a role as a minor fetal excretory pathway, particularly during the second trimester. Experiments using nitrous oxide in primates provide support for this excretory mechanism.79 The recent demonstration of a sizable tidal flow of amniotic fluid through the fetal lungs in relation to fetal respiratory movements during the last trimester raises the intriguing possibility of drug exchange at this level.80 Finally, fetal swallowing may allow certain drugs to be recirculated. The early maturation of the intestinal enzyme glucuronidase may make it possible to hydrolyze conjugated compounds excreted by the fetal kidney and permit their intestinal reabsorption.
Pharmacologic and toxic effects in the fetus
Drugs exert their effect at receptor sites. Information concerning the pattern of development of drug receptors during fetal life remains sketchy, fragmentary, and for the most part unknown. From studies of the functional development of individual organs in the fetus, it can be deduced that maturation of pharmacologic receptors is likely to proceed at different rates. With the notable exception of the antepartum administration of glucocorticoids for the prevention of hyaline membrane disease or treatment of fetal cardiac arrhythmias, pharmacologic manipulation of the fetus remains a hope for the future. Our present concerns deal with toxic and teratologic effects. There are three successive periods in fetal development for drug-related teratologic and toxic effects: (1) fertilization and implantation (days 0–17), at this stage a fetotoxic agent can cause death of the embryo; (2) organogenesis (days 18–55), this is the most sensitive period for development of malformations; and (3) fetal period (56 days to birth); in this stage drugs can decrease cell size and number or affect the organization of the cerebral cortical layers.
Drugs may affect the fetus directly or indirectly by changes in maternal nutrient delivery to the fetus, changes in placental circulation, or alterations in maternal glucose homeostasis. Both acute and chronic drug exposures can cause adverse effects.
Acute toxicity occurs mostly during labor and delivery, when the fetus abandons its major drug excretory organ, the placenta. Rarely has fetal poisoning been the result of attempted suicide in the mother, ingestion of toxins unknown to the mother, or mistaken medications. The overwhelming concern, however, centers on the effect of drugs and environmental chemicals during pregnancy. Distinction between toxic and teratologic effects seems arbitrary and unwarranted. Traditionally, teratologic effects have been defined as congenital anatomic malformations secondary to deleterious agents administered during the first trimester of pregnancy. This definition is misleading and too confining. It is now widely acknowledged that drugs and other agents taken at any time during gestation can produce different types of developmental defects, including prenatal or postnatal growth retardation, distortion of cell architecture of the cerebral cortex, postnatal functional or behavioral disorders, and prenatal or postnatal neoplasms. The concept of developmental toxicity is preferred because it is more encompassing, does not presuppose the presence of anatomic defects, and allows consideration of functional abnormalities produced by the exposure. In fact, drug effects may not be seen until many years after birth (e.g., adenocarcinoma of the vagina after intrauterine exposure to diethylstilbestrol).
The fear of fetal toxicity often results in undertreatment, which in turn may complicate the interpretation of an abnormal outcome. It is known that chronic exposure to tricyclic antidepressants may lead to fetal growth deficits. Likewise, women with symptoms of depression have an increased risk of delivering a low-birth weight infant.
A similar situation occurs with the treatment of chronic hypertension. Prematurity and low birth weight may result from the placental vascular changes associated with hypertension or may result from chronic exposure to some antihypertensive drugs.
A number of factors complicate the interpretation of the available data on the fetal consequences of drug exposure during pregnancy. The determination of drug exposure by retrospective recall is known to be associated with a high error rate. Multiple drug exposures are often not recorded; drug–drug interactions are difficult to determine; there is an overreliance on the clinical significance and the predictive value of animal studies; and there is a paucity of long-term studies.
Obstetric-fetal pharmacology in the genomic era
Recent advances in genomic technology and health information have led to an expansion of pharmacogenetics and a move toward personalized medicine. The goal of tailoring drug dosage to an individual's unique genetic make-up, instead of a trial-and-error and one-size-fits-all approach, is beginning to be realized. The application of personalized medicine in obstetrics is complicated by the need to take into account the genetic makeup of both mother and fetus. Clinically, relevant polymorphisms of drug metabolizing enzymes affect the disposition of specific drugs similarly in pregnant and non-pregnant women. While genetic polymorphisms of several transport proteins and drug receptors have been described, their relevance in practice remains unexplored. Also unknown is the role of the pregnant state on genetic expression of drug effectors. The interests of mother and fetus are often divergent as most drugs are given to benefit the mother. Efficacy and low toxicity is paramount for the mother and lack of toxicity is the fetal priority. In the fetus, it is often difficult to differentiate ontogenic versus genetic factors (e.g., CYP2D6 activity is low in the fetus for both fast and slow metabolizers because genotype will not manifest until several months after birth). Targeting therapy to genotype implies that knowledge of the efficacy/toxicity profile for each drug is known according to genotype. In fact recommendations for drug use according to genotype have been established for few drugs (e.g., psychotropic drugs, antiasthma drugs) and only in non-pregnant patients. Although, it is too early to foresee the immediate implications of pharmacogenomics in obstetrics, advances in genomics will have an immediate application in the determination of drug responses and determination of toxicity.81
PREGNANCY RATINGS AND DRUG LABELS
In 1979, the FDA developed a classification system intended to provide guidance to physicians considering prescribing drugs to pregnant women. Drugs are allocated to one of the following categories:
- Category A : No risk to the fetus has been demonstrated, and there are adequate and well-controlled studies in humans.
- Category B: There is no evidence of risk in human studies and animal studies are either positive or lacking.
- Category C: Human studies are lacking and animal studies are positive or lacking.
- Category D: Positive evidence of human fetal risk based on adverse reaction data, but the potential benefits outweigh the potential risk.
- Category X: Animal data or human data indicate fetal anomalies, and the risk of use clearly outweighs its benefits.
A review by FDA Pregnancy Labeling Task Force of 1213 drug labels listed in the Physicians' Desk Reference revealed the following frequency distribution: category A, six drugs (five thyroid hormone supplements and one iron/folic acid supplement); category B, 228 drugs; category C, 806 drugs; category D, 83 drugs; and category X, 90 drugs. Analysis of the information revealed a number of inconsistencies. Studies were not done in 49% of drugs in category C and in 60% of drugs in category X; 77% of drugs in category D did not have human data. More revealing is the analysis of toxicity and teratologic data. In category D, toxic effects were listed in 11%; 8% of drugs were teratogenic and in 4% toxic effects included evidence of teratogenicity. In category X minimal effects were listed in 13% of the drugs reviewed, toxic effects in 6%, and teratogenic effects in 13%. Toxic effects, including evidence of teratogenicity, were listed in 8% of the drugs in category X. Other problems with the pregnancy ratings included incomplete or erroneous information, incorrect category assignments, and lack of updated information.
The FDA has revised the labeling system in pregnancy and added a section on "Lactation Labeling". The development of a system that provides a realistic estimate of fetal risk from human studies and is less reliant on animal data is a major step forward.
Under the proposed rule the subsection entitled "Labor and Delivery" would be eliminated and the pertinent information would be included in the proposed “Pregnancy” section.82 If adopted the proposed rule will eliminate the current five categories. Both the "Pregnancy" and "Lactation Labeling" would include three headings: risk summary, clinical considerations, and data. The fetal risk summary would emphasize human data. The overall risk conclusion would state if the conclusion was based on human or animal data. When human data are available the most significant data about the effects of the drug on the fetus will be included. Additionally, under the proposed rule, the pregnancy subsection of the labeling would be required to include information on any pregnancy registry that had been established for the drug.
USE OF PHARMACOKINETICS AND PHARMACODYNAMICS MODELS IN PREGNANCY
The time course of exposure to drugs in pregnancy has revealed pharmacokinetic alterations for several drugs given to pregnant women.83 Unfortunately, the extant pharmacokinetic data for most drugs given to pregnant women are not generalizable because of insufficient design, small sample size, use of unadjusted maternal weight values, and lack of longitudinal studies across different gestational ages. Reference standards have varied with different comparison groups: nonpregnant women, historical controls (mostly nonpregnant adults), adult men, and the same patients studied in their postpartum period. The methods used to determine gestational age were also not uniform. The pharmacokinetics studies available have not been stratified according to maternal condition (normal and abnormal pregnancies). There is a dearth of bioavailability studies.
Although the current knowledge of pharmacodynamics in pregnancy is quite limited, there is evidence of changes in drug sensitivity for some drugs in comparison with the nonpregnant state.
The current approach for the study of pharmacokinetics and pharmacodynamics is the use of mathematically based models that focus on the mechanisms of drug action. Jusko and coworkers84 described the components of pharmacokinetic/pharmacodynamic models (Fig. 2). Several steps need to be considered depending on the mechanisms of drug action between changes in drug plasma concentrations and measured effects.
|
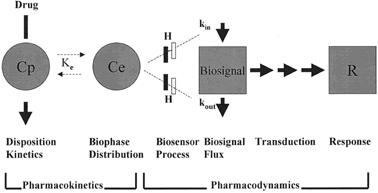
For some drugs, a biphase or effect compartment is included to estimate drug concentrations at the site of pharmacologic effect. A number of drugs exhibit delayed responses because they act by indirect mechanisms (e.g., stimulation or inhibition of mediators or receptor-mediated process). Turnover of mediators or bound receptors represents biosignal flux. Pharmacologic effects may utilize a signal transduction process. This cascade of events is the result of interactions between drug molecules and receptors with or without the involvement of second messengers. The signal transduction pharmacokinetic-pharmacodynamic (pk-pd) model estimates the signal transduction process that produces the drug effect or response. Wyska and Jusko85 have applied these pk-pd models to data available in the literature. The results of this pk-pd evaluation comparing pregnant and nonpregnant subjects revealed differences in clotting response to heparin, decreased sensitivity to labetalol, and changes in recovery times after the intravenous administration of succinylcholine during pregnancy.
There is also experimental evidence of specific receptor alterations during pregnancy.86 A paradoxical relationship between pharmacokinetic and pharmacodynamic response to vecuronium has been reported. The effect of the drug increased 78% during pregnancy, whereas its half-life was considerable decreased.87 A pk-pd correlation study of metoprolol during pregnancy revealed a minimal difference in response: changes in blood pressure coincided with a fourfold decrease in the plasma drug concentration.88 These observations illustrate the need to perform pk-pd correlation studies and the potential danger of making dose adjustments in pregnancy without knowing the sensitivity of the drug. Pharmacokinetic models can also be used to relate maternal and fetal drug concentrations and to determine fetal drug exposure.
There is a voluminous literature on maternal and newborn drug concentration ratios at birth for different drugs. The newborn data consist of a single-point determination obtained under nonsteady-state conditions; placental circulatory changes associated with labor further complicate the interpretation of maternal–fetal gradients and the estimation of fetal drug exposure.
The inaccessibility of the human fetus makes it necessary to use animal models, and species differences may limit the applicability of pharmacokinetic data to humans. It is theoretically possible to use primate animal models to develop pharmacokinetic profiles and relate them to the available information on maternal–fetal drug concentrations.
EFFECT OF PREGNANCY ON SPECIFIC CONDITIONS: PHARMACOLOGIC IMPLICATIONS
The natural history of pre-existing conditions may be altered by pregnancy, and changes in the natural history of diseases may in turn affect response to therapy.
Between one quarter and one third of women with epilepsy experience an increase in seizure frequency during pregnancy.89 This increase appears to be unrelated to the type of seizure, the duration of epilepsy, and the frequency of seizures in previous pregnancies.
A protective role of pregnancy has been advocated for some conditions, but systematic studies are lacking. The commonly held view that the rates of depression during pregnancy are lower than in the nonpregnant state has been recently challenged.90 In contrast, the increased risk of depression during the postpartum period is universally acknowledged. The course of chronic or essential hypertension may be influenced by pregnancy. The marked hemodynamic changes and the increase in blood volume associated with pregnancy may have a detrimental effect on pre-existing cardiovascular disease.
The risk of derangement in carbohydrate metabolism is increased during pregnancy. These alterations may lead to higher insulin requirements in women with insulin-dependent diabetes.
The anatomic and functional changes in the respiratory system associated with pregnancy may affect the course and response to therapy in respiratory illnesses. The changes in the immune system during pregnancy have been implicated as a cause for worsening of asthma. The effect of pregnancy on the natural history of asthma, however, remains controversial. The increase in circulating hormones such as progesterone and aldosterone, which are known to be competitive antagonists for glucocorticosteroid receptors, may reduce the response to oral or inhaled steroids.91
Acute pyelonephritis complicating untreated bacteriuria is associated in a significant number of patients with a marked reduction in creatinine clearance.92,93 This reduction in renal function can have a significant effect on the pharmacokinetics of drugs excreted primarily by the kidneys.
SPECIFIC DRUGS: CLINICAL CONSIDERATIONS
Data on pharmacokinetics during pregnancy are extremely scarce and often contradictory. Investigators may conclude that adjustments in doses are necessary but stop short of making specific recommendations. An obstetrician or subspecialist can find very little reassurance from reading the literature on specific drugs as to the most appropriate dose and treatment schedule for his or her patient.
It has been generally assumed that drugs proven to be effective in nonpregnant patients are equally effective in pregnant patients. Evidence of pharmacokinetic and pharmacodynamic changes for certain drugs has been noted above. Similarly, a change in the course of disease during pregnancy may also alter the efficacy of drugs. In theory, randomized control trials could be used to evaluate the efficacy of any drug given during pregnancy. It is ethically unacceptable, however, to use pregnant women in a placebo group, with the possible exception of life-threatening conditions for which there is no effective treatment. Longitudinal studies that objectively measure outcomes in the same women during pregnancy and in the postpartum period are an acceptable study design.
No attempt has been made to review all drugs known to exert adverse effects on the fetus or neonate. Instead, specific drugs have been selected according to frequency of use and availability of data in humans. Fetal effects are included, but discussion of the relative risk is beyond the scope of this review.
Absence of demonstrable effect does not absolve a drug, because toxic manifestations of intrauterine exposure may be subtle, unexpected, and delayed. There are serious gaps in our knowledge because often adverse effects are identified at the functional or structural level. Unraveling of the biochemical or metabolic basis for the observed effects is fundamental if preventive methods are to be developed. On the other hand, drugs may be given to the mother to treat the fetus (e.g., corticosteroids to induce lung maturation). Similar pharmacokinetic and pharmacologic considerations apply as in dose-related adverse effects, but at different dosages.
DRUGS GIVEN DURING PREGNANCY
Cardiovascular drugs
Cardiac glycosides are used in pregnancy to treat heart failure and to manage fetal tachyarrhythmias. The pharmacokinetics of digoxin was studied during the third trimester and again in the postpartum period.56 The puzzling observation was made that the higher digoxin renal clearance, creatinine clearance, and 24-hour urine elimination of digoxin were associated with higher serum digoxin concentrations during the third trimester compared to the corresponding postpartum values. Recent work has shown an endogenous digoxin-like substance produced by the placenta or fetus that interferes with the radioimmunoassay of digoxin. Measurable amounts of this substance may explain the high digoxin levels found in the third trimester.94 Concern has been expressed that moderate- to high-dose intrauterine infusions of dopamine, dobutamine, and norepinephrine may harm the fetus because of decreased uterine blood flow or premature stimulation of uterine contractions.95 Limited information is available on the use of quinidine, procainamide, lidocaine, disopyramide, amiodarone, verapamil, and other antiarrhythmic drugs during pregnancy. Quinidine has been used extensively to treat supraventricular and ventricular tachycardia in the mother and rarely for the termination of fetal tachyarrhythmias. No significant fetal side effects have been reported. The pharmacokinetics of antiarrhythmics has been reviewed.96 Propranolol and other beta-adrenergic blocking agents have been used for the treatment of atrial arrhythmias, hypertrophic obstructive cardiomyopathy, and hyperthyroidism. The relative risk of fetal growth retardation associated with the prolonged use of propranolol remains to be determined.
Antihypertensive medications
The major therapeutic goal of treating hypertension in pregnancy is to protect the mother from the development of acute complications and to deliver a healthy infant.97 There is no convincing evidence that treatment of mild or moderate hypertension prevents or delays the development of preeclampsia. Antihypertensive drugs are widely used to treat chronic hypertension, gestational hypertension, or preeclampsia. The accurate measurement of blood pressure is essential and the diagnosis of hypertension or preeclampsia cannot be based on a single measurement. It is important to stress that a recent study found white coat hypertension to be common among pregnant women.98 The indications and the benefits of different antihypertensive drugs in pregnancy have been recently reviewed.99, 100 Methyldopa, beta-blockers, calcium channel blockers, and hydralazine have been used for all forms of hypertension. Most randomized controlled clinical trials of antihypertensive drugs in patients with mild hypertension or preeclampsia had inadequate power to demonstrate efficacy. Similarly, comparative trials between labetalol, nifedipine, and hydralazine were not sufficiently powered to demonstrate superiority or even equivalence between treatments.99, 101 There is a dearth of information on the pharmacokinetics and pharmacodynamics of most antihypertensive drugs. There is a lack of consistency in pharmacokinetic data on specific antihypertensives or between different types of antihypertensives. In one study, an increased clearance of labetalol was demonstrated, whereas two other studies failed to show differences between pregnant and nonpregnant patients.101, 102, 103 Plasma concentration, area under the curve, and half-life of metoprolol were found to be decreased during the third trimester. From the pharmacologic standpoint, hypertension in severe preeclampsia should be considered a manifestation of a complex syndrome.
Longitudinal studies are needed, particularly in the second and third trimesters, to determine the pharmacokinetic consequences of the reduced plasma volume, decrease in protein binding, reduced hepatic metabolism, or alterations in renal function associated with severe preeclampsia.
The unraveling of the role of mediators involved in the pathogenesis of severe preeclampsia may lead to a targeted pharmacologic approach. For example, it has been suggested that serotonin may play an important role in preeclampsia. The concentration of platelet-derived circulating serotonin is significantly increased in women with preeclampsia.104, 105, 106 The use of a selective serotonin receptor antagonist (ketanserin) has been proposed for the treatment of hypertension in preeclampsia. Few clinical trials have so far been conducted using chronic ketanserin therapy.107 A mild prolongation of the QTc interval has been found in a few treated patients. It remains for future research to determine whether a targeted pharmacologic approach in toxemia in preeclampsia is more efficacious and safer than conventional treatment.
All drugs given to pregnant women for the treatment of hypertension can be expected to cross the placenta and reach significant concentrations in fetal blood. The potential adverse effects for most commonly prescribed antihypertensive drugs have not been adequately quantified. Alpha-methyldopa, an amino acid analog, uses the same transport mechanism as vitamins and amino acids and is an effective antihypertensive. Its popularity in nonobstetric practice has waned in recent years because of unwanted side effects, but its unique safety for the newborn has been established extensively in obstetric literature. Hydralazine remains the most commonly used antihypertensive for the acute treatment of severe toxemic states. This drug occasionally produces hypotension and acute uteroplacental insufficiency resulting in fetal distress. The NHLBI guidelines for the treatment of antihypertension in pregnancy recommend methydopa as a first-line therapy because of its safety for the fetus; oral labetalol is an alternative. Beta-blockers and calcium antagonists are considered acceptable by the committee.108
Angiotensin-converting enzyme and ACE inhibitors administered during pregnancy may cause lethal renal failure at birth. Magnesium sulfate, the most commonly used drug for the treatment of preeclampsia and eclampsia, is considered ineffective as an antihypertensive agent.
Randomized or controlled studies of the use of beta-blockers (e.g., atenolol, metoprolol, and oxprenolol) in the management of pregnancy hypertension have vindicated these agents from the pessimistic view of a high incidence of fetal side effects gathered from anecdotal reports or retrospective studies.109 The frequency of hypotension, bradycardia, and respiratory problems was found to be less significant than previously anticipated. With the exception of propanolol, hypoglycemia was not found to be a problem. There remains a concern, heightened by experimental evidence, that maternal adrenoreceptor blockade may reduce fetal tolerance to asphyxia. Labetalol, a nonselective beta-blocker with some alpha-1 blocking activity, is being evaluated. Labetalol appears to be an effective and seemingly safe alternative to hydralazine for the treatment of hypertension in the peripartum period.110 To date there are no convincing studies showing the superiority of labetalol. One study compared labetalol with methyldopa in a randomized controlled trial which enrolled 176 pregnant women. Blood pressure reduction, heart rate, and average birth weight was similar in both groups.111 In another trial comparing the two drugs, the labetalol group achieved a faster and more efficient blood pressure control.112 Practicing obstetricians continue to rely on methyldopa because of its well established safety record. Labetalol remains a second choice.
A large case–control study of birth defects in early pregnancy showed an increased risk of severe hypospadias for untreated maternal hypertension and an association between hypertension, antihypertension medications and the risk of severe hypospadias.113 The significance of this finding and the possible etiologic mechanisms involved remain undetermined.
It is possible that antihypertensive drugs, known to interact with brain neurotransmitters and thus to lower maternal blood pressure, may interfere with the development of brain transmitters in the fetus. This possibility has not been adequately explored.
Pulmonary pharmacology
The absorption of drugs administered by inhalation may be influenced by the physiologic changes that occur during pregnancy. The increased pulmonary blood flow, coupled with hyperventilation, may increase the absorption of inhalants.114 This possibility has not been documented by systematic studies. The short-term inhalation of a maximum dose of albuterol did not affect the fetal heart rate or aortic velocities in the fetus.115
Bronchial asthma occurs in 4–8% of pregnancies. Acute asthma is potentially dangerous to the fetus. Juniper and Newhouse116 performed a meta-analysis of studies researching the effect of pregnancy on the course of asthma and found that in one third of the affected women, the disease worsened. More importantly, pregnant women with severe asthma are more prone to have a complicated course in pregnancy.
Severe and uncontrolled asthma during pregnancy may cause serious maternal and fetal complications such as gestational hypertension and eclampsia and an increased risk of perinatal death. In addition inadequate control of asthma during pregnancy is associated with a significant risk of preterm delivery.117
Corticosteroids facilitate the effect of beta-agonists on cAMP production and decrease the activity of inflammatory cells. Beclomethasone has been proposed as the inhalational steroid of choice for the treatment of asthma in pregnancy.116 There are no pharmacokinetic or pharmacodynamic studies during pregnancy on oral or inhaled steroids. It has been suggested that circulating progesterone, aldosterone, and deoxycorticosterone, which are competitive antagonists of glucocorticoid receptors, could reduce the sensitivity to oral or inhaled steroids during pregnancy,118 but to date no studies have addressed this possibility.
Human data dealing with the safety of drugs during pregnancy are usually limited to observational studies. There is one randomized controlled study supporting the efficacy of inhaled beclomethasone for the treatment of asthma during pregnancy.119 Other therapeutic options such as cromolyn and nedocromil deserve consideration as alternatives to inhaled corticosteroids. In cases of severe persistent asthma add-on therapy (e.g., leukotriene receptor antagonists, long-acting beta 2-adrenergic agonists) should be considered.120 No significant adverse effects or congenital malformations have been reported with maternal inhaled steroid exposure. In contrast, a meta-analysis of published studies on the use of systemic steroids during the first trimester of pregnancy revealed an increased risk of oral clefts.121
Pharmacologic treatment of gestational diabetes
Insulin has been the traditional treatment for gestational diabetes unresponsive to dietary control.122 The worldwide increase in the incidence of type 2 diabetes, has stimulated the development of newer antidiabetic drugs that are more effective or have a better risk–benefit ratio than the first generation sulfonylureas. However, concerns over the teratogenicity of oral antidiabetic drugs have limited their use in pregnancy.123 Animal studies have found no evidence of teratogenicity of glyburide, glipzide, metformin or rosiglitazone. Unfortunately, human information is inadequate or not available for many of these compounds. Safety of antidiabetic drugs in pregnancy must include evidence of lack harmful toxic effects (e.g., hypoglycemia, macrosomia) in the fetus. Transplacental transfer is a major determinant of the fetal toxicity of antidiabetic agents. The insulin analog insulin Lispro does not cross the placenta because of its large molecular weight. The breast cancer resistant protein protects the fetus by blocking glyburide penetration across the placenta.124 A recent review of randomized controlled trials and observational studies of maternal and neonatal outcomes treated with oral antidiabetic drugs compared with insulin found four randomized and five observational studies.125 No significant difference in glycolic control or cesarian deliveries was found in the randomized trials (two trials compared insulin to glyburide, one trial compared insulin, glyburide, and acarbose and one trial compared insulin to metformin. Despite studies showing a high success rate of glyburide, there are reports of glyburide-associated macrosomia, neonatal hypoglycemia, and increased risk of preeclampsia.126 Further research is needed before glyburide and other newer oral antidiabetic drugs can be recommended for routine use in pregnancy.
Psychopharmacologic drugs
Psychopharmacologic drugs are commonly used as sedatives and for the treatment of psychiatric conditions. It has been estimated that 35% of pregnant women receive psychotropic drugs.127 As with other drugs used in pregnancy, there is a lack of pharmacokinetic and efficacy studies for most psychotropic drugs.
Lithium is used for the treatment of bipolar states. During pregnancy there is a decrease in the plasma lithium concentration compared with the nonpregnant state. Lithium carbonate readily crosses the placenta. Marked cyanosis and hypotonia are signs of intoxication at birth. Lithium inhibits thyroid hormone secretion. In adults, goiter is a well-known side effect of lithium carbonate therapy. Goiter and transient hypothyroidism have been infrequently reported in neonates as a consequence of fetal exposure to lithium carbonate.128 Nephrogenic diabetes insipidus, a common side effect in adults treated with lithium, has also been described in infants whose mothers were treated with this medication.129 The use of lithium during pregnancy has been associated with perinatal complications and reversible neonatal toxicity including depressed neurological status, hypotonia, and lethargy.130
Clorazepate, a drug used for the treatment of anxiety, has been studied during the last trimester of pregnancy and after delivery. The drug plasma concentration and half-life were found to decrease and clearance was found to increase at the end of pregnancy.131 There is limited information on the pharmacokinetics of antidepressants during pregnancy. Protein binding of tricyclic antidepressants, benzodiazepines, and neuroleptics is decreased, with a consequent increase in the free fraction of the drug. At standard doses, the blood concentrations of clomipramine and imipramine decrease during pregnancy.132
There are no data on the effects of pregnancy on the plasma concentration of selective serotonin reuptake inhibitors (SSRIs), bupropion, trazodone, or sertraline. It is an unfortunate practice to reduce the dosage or even discontinue antidepressants on learning that a patient is pregnant. This practice increases the likelihood of a relapse and sometimes leads to suicide.
Exposure to SSRIs in late gestation may result in clinical manifestations including neurobehavioral, respiratory, gastrointestinal, and persistence of pulmonary hypertension syndrome in as many as 30% of neonates.133 Recent studies have shown increased depressive symptoms in late pregnancy, requiring dose increases to achieve remission.134 A number of isolated case reports have documented that adverse fetal effects may occur with the maternal administration of psychotropic drugs. Withdrawal symptoms have been reported in newborns exposed in utero to drugs such as desmethylimipramine, ethchlorvynol, pentazocine, and chlordiazepoxide.135, 136, 137 The variability in the onset, severity, and duration of symptoms can be ascribed to many factors, including the type of drug, maternal dosage, fetal accumulation, and patterns of drug elimination after birth. The limited ability of the newborn to metabolize and eliminate these drugs may be responsible for the fact that withdrawal symptoms may be significantly delayed for 2–4 weeks. The clinical symptomatology cannot be distinguished from that of infants undergoing narcotic withdrawal. Tremors, irritability, a high-pitched cry, hypertonicity, and, on occasions, loose stools and vomiting have been observed.
Withdrawal signs from phenothiazines feature extrapyramidal signs, including opisthotonus, hypertonia, and hand posturing, which persist for several months in some infants. Tricyclic antidepressants are slowly metabolized by the fetus and newborn; withdrawal symptoms include heart failure, tachycardia, myoclonus, and urinary retention. The risks and benefits of psychotropic drugs have been reviewed.138
Anticonvulsant medications
Management of epilepsy during pregnancy constitutes a therapeutic challenge. The goal is to keep the patient seizure-free while minimizing the adverse effects of antiepileptic drugs on the fetus.139 The plasma concentrations of antiepileptic drugs (AEDs) decrease during pregnancy. The percentage decreases in total AED plasma concentrations in the third trimester for carbamazepine, phenytoin, phenobarbital, and ethosuximide are 40%, 56%, 55%, and 90%, respectively. Several mechanisms may be responsible for this decline: intestinal malabsorption, decreased plasma protein binding (particularly to albumin), increased drug metabolism, and induction of metabolizing enzymes by comedications.140, 141 The plasma concentrations of the different AEDs decline at different rates throughout pregnancy but consistently return to nonpregnant levels in the postpartum period. Since 1993, eight new and effective AEDs have been approved for use in adults: felbamate, gabapentin, lamotrigine, oxcarbazepine, topiramate, tiagabine, trileptal, and zonisamide. Scanty information is available on the effectiveness and safety profile of these newer AEDs during pregnancy.
Numerous reports have highlighted the teratogenic effects of phenytoin and other AEDs.142, 143 Infants exposed to AEDs in utero are twice as likely to develop birth defects as those not exposed to these compounds. The risk of congenital malformations, including neural tube defects, after exposure to the new AEDs is not known.144 The fetal hydantoin syndrome consists of unusual facies, digit and nail hypoplasia, and growth and mental retardation, along with other congenital anomalies. This syndrome has been linked with the development of neural crest tumors.145 Age at presentation of the neoplasms has varied between 1 day and 36 months.
Pregnancy registries set up to obtain information about the potential risks of fetal exposure to AEDs, in particular major congenital malformations (MCMs), suggest that valproate exposure increases the frequency of congenital malformations more than other AEDs. Furthermore, follow-up studies have drawn attention to cognitive impairments in later childhood after prenatal exposure to valproate.146
Folate deficiency during pregnancy has been widely documented in women taking anticonvulsants; its relationship to adverse pregnancy outcome remains to be determined.147, 148 Fetuses exposed to AEDs are known to develop bleeding tendencies during the first day of life. This coagulopathy, initially reported after exposure to phenobarbital, has also been described in newborns exposed to phenytoin, carbamazepine, diazepam, mephobarbital, amobarbital, and ethosuximide. It would appear that anticonvulsants antagonize the action of vitamin K by preventing the hepatic gamma carboxylation of the vitamin K-dependent coagulation factors: II (prothrombin), VII, IX, and X.149 In vitamin K-deficient states, an abnormal type of prothrombin named protein-induced vitamin K absence factor (PIVKA II) is produced. In the coagulopathy induced by AEDs, the maternal concentration of coagulation factors remains normal; the fetus has a decrease in the above-mentioned clotting factors and an increase in PIVKA II. Despite the decrease in clotting factors, the fetus receives enough maternal vitamin K in utero. After birth, exogenous sources must be provided.
Fetal exposure to AEDs may be influenced by drug transporting proteins in the placenta, including P-glycoprotein (P-gp), multidrug resistance protein (MRP) 1, and breast cancer resistance protein (BCRP).150 Genetic variations in the expression and activity of these transport proteins may influence fetal exposure to AEDs and thus the risk of teratogenicity. Identification of a hierarchy of haplotypes ranging from susceptible to protective of congenital abnormalities could assist genetic counseling, in assessing fetal risks from exposure to AEDs.
The rational use of AEDs during pregnancy requires knowledge of the pharmacokinetic characteristics through establishment of pk-pd correlations, routes of administration, and the effect of coadministering competing drugs. The gaps in current knowledge allow only tentative recommendations to be made. Use of single-drug therapy is preferable whenever possible. The drug of choice is the one that provides optimal seizure control with minimal side effects. With the possible exception of trimethadione, the risk of teratogenicity is relatively small. Overestimating this risk may be in itself an even greater risk if one considers the outcome of poorly controlled epilepsy.
Plasma drug levels should be frequently monitored because of the increased requirements during pregnancy. This latter factor is particularly significant with phenytoin during the third trimester. Measurement of total plasma phenytoin levels during pregnancy does not reflect the amount of pharmacologically active free drug and is therefore inadequate. Saliva phenytoin measurements correlate closely with free phenytoin levels in plasma and offer a practical solution to the problem of estimating therapeutic concentrations.151 The doses needed for maintenance often reach amounts that could be considered toxic in the nonpregnant state.
Status epilepticus should be approached with the same therapeutic conviction in the pregnant patient as in other adult patients. Because of inadequate oxygenation and circulation, timidity in prescribing constitutes a bigger threat to the fetus than the untoward effects that may be associated with rapid intravenous loading doses of anticonvulsants.
Antineoplastic agents and immunosuppressant agents
Because of improvements in the effectiveness of therapy for pediatric malignancies, an increasing number of girls with cancer are reaching reproductive age. The use of antineoplastic drugs may be associated with premature delivery. The association may be coincidental because premature birth may be due to poor nutrition, overt metastatic disease, and possible prior abdominal irradiation. A study of 33 offspring of women who received abdominal irradiation for Wilms tumor showed a 21% incidence of prematurity.152 The major concern regarding the use of antineoplastic agents in pregnancy, however, is their teratogenic potential. Teratogenic risk is related to several factors, including whether single- or multiple-drug regimens are used, the time of exposure, and the class of antineoplastic agents used. Antineoplastic drugs as a group comprise several chemically different and unrelated compounds (e.g., antimetabolites, alkylating agents, spindle poisons, antibiotics, and hormones). The demonstration of embryotoxic effects in pregnant hospital personnel exposed to antineoplastic agents is of considerable interest and indicates the need for appropriate precautions.153 It has been estimated that fewer than 1 in 70,000 pregnancies is complicated by leukemia.154 The question of what to do with a pregnancy when the diagnosis of leukemia is established cannot be easily answered. It is generally accepted that therapeutic abortion should be recommended in patients requiring chemotherapy during the first trimester of pregnancy.
Chemotherapy for acute leukemia during the second trimester is controversial. Pregnancy may be allowed to continue if the mother is willing to accept the unknown risk of fetal anomalies. The type and combination of drugs, timing within the second trimester, and variety of leukemia will influence fetal outcome. It is generally conceded that chemotherapy is relatively safe if treatment is started during the last trimester of pregnancy. Certain drugs, such as alkylating agents and procarbazine, should be avoided at all stages of pregnancy.
The use of cyclophosphamide is associated with an increased therapeutic risk, but the magnitude of the risk remains undefined. There are serious gaps in our knowledge in this area. The above recommendations are based on a rather small number of patients.154, 155, 156, 157, 158, 159 Potential delayed toxicity, risks of leukemia in childhood, and acquired genetic disorders are concerns that have not been adequately explored.
Pregnancy associated breast cancer is increasing in frequency and is estimated to be the second most frequent malignancy in obstetrics. There are few limitations in treatment choices, except for radiotherapy, and hormonal of antibody treatment. Treatment recommendations are based on nonrandomized studies mostly case reports and anecdotal series of patients.160
The increased survival of organ transplant recipients raises concern about the fetal effects of immunosuppressant drugs. Infants born to women who received renal transplants have perinatal complications including intrauterine growth retardation, marrow hypoplasia with leukopenia, and thrombocytopenia. It is not clear whether these complications are due to the toxic effects of azathioprine or chronic immunosuppression.161
Antituberculous drugs
Tuberculosis is more aggressive during pregnancy, particularly in HIV-positive women. HIV co-infection results in greater mortality in developing countries.162
The potential teratogenicity of antituberculosis compounds during pregnancy has been reviewed.163 Setting aside concerns over possible embryotoxic effects, the so-called first-line drugs (i.e., isoniazid, ethambutol, and rifampin) seem to be relatively safe. The most commonly used regimens whenever a tuberculous infection is detected are isoniazid, ethambutol, isoniazid plus rifampin, or isoniazid plus ethambutol plus rifampin. Pooling data from different investigators, the survey uncovered a remarkable margin of safety for isoniazid: more than 95% of 1480 pregnancies studied resulted in the birth of a normal term infant.
About 1% of the infants were noted to be abnormal; it is not clear from the report whether those abnormalities were related to the well-known central nervous system toxicity of the drug. Isoniazid readily crosses the placenta and may achieve fetal plasma concentrations higher than those in the maternal plasma. When isoniazid is used, supplemental pyridoxine is recommended because of the increased requirement for this vitamin during pregnancy. Streptomycin ototoxicity was found in an unexpected large number of fetuses exposed to the drug: one in six of the fetuses exhibited some degree of hearing loss or vestibular dysfunction. The authors emphasized that streptomycin ototoxicity is not confined to the period of embryogenesis because it can occur at any time during pregnancy. Ethambutol was found to be safe, but the relatively small number of rifampin pregnancies studied precluded a definite pronouncement about its safety.
Occasionally, because of resistance, drugs other than the primary agents cited may be needed. Unfortunately, there is no available information on the pharmacokinetics of para-aminosalicylic acid, viomycin, or capreomycin. Further, their use is limited mostly because of potential side effects on the mother rather than for concern of untoward effects on the fetus.
Ethionamide is considered contraindicated in pregnancy because of its teratogenic effect.164
Anticoagulants
The treatment of thrombophlebitis and other embolic disorders during pregnancy poses a special problem in the choice of anticoagulants that are safe for the fetus. Heparin is the anticoagulant most commonly used because of its large molecular weight, ranging from 4000 to 40,000 daltons; only an insignificant amount of this drug cross the placenta, and therefore, it does not affect the fetus. Disadvantages of heparin include the variability of biologic activity of commercial preparations, the need for parenteral administration, and the difficulty in monitoring bioeffect.
Pregnancy produces an increase in the concentration of coagulation factors, particularly fibrinogen and factor VIII.165 The physiologic increase in the levels of coagulation factors can produce a shortening of the activated partial thromboplastin time (APTT) compared to nonpregnant controls. In addition, nonspecific plasma protein binding increases in pregnancy. The binding of high-molecular-weight chains of unfractionated heparin to plasma protein results in lower plasma heparin measurable levels compared to nonpregnant women.166 A corollary of these findings is the potential of overdosing when the heparin dosage is based on APTT standards based on the nonpregnant therapeutic range.
Adverse maternal effects of unfractionated heparin therapy are thrombocytopenia, hypersensitivity, and osteopenia. The incidence of heparin-induced osteopenia is low.167
Although bone loss associated with heparin exposure in pregnancy may be partially reversed a year after therapy, it is unknown whether it can contribute to the risk of osteoporosis later in life.168
Low-molecular-weight heparin (LMWH) has several advantages over unfractionated heparin. LMWH binds less readily to plasma protein compared to unfractionated heparin. LMWH also has a more predictable dose–response and half-life.169 The pharmacokinetics of LMWH during pregnancy has not been adequately studied. LMWH distributes in plasma and is excreted by the kidneys. Physiologic changes such as an increased glomerular filtration rate and an increase in plasma volume may alter the biodisposition of LMWH during pregnancy. It has been shown that LMWH given at prophylactic doses results in peak and trough heparin levels, measured by antifactor Xa activity, that are lower during pregnancy when compared to the nonpregnant state.170
Current evidence indicates that LMWH does not increase the risk of maternal bleeding provided that the drug is discontinued at least 12 hours prior to delivery to prevent postpartum bleeding. The incidence of LMWH appears low compared to unfractionated heparin. Fondaparanux or danaparoid may offer an alternative to the use of unfractionated heparin in pregnant women with heparin-induced thrombocytopenia.171
Warfarin, with a molecular weight of 1000, crosses the placenta and profoundly affects the fetal prothrombin time. Pregnant women receiving warfarin in early pregnancy because of prosthetic heart valves are at risk to give birth to deformed children. The fetal warfarin embryopathy associated with exposure during the first trimester of pregnancy includes abnormal facies (hypoplastic nose and depressed nasal bridge), stippled epiphysis and vertebrae, extremity shortening, central nervous system dysfunction (including mental retardation), and eye abnormalities. Exposure only during the second and third trimesters is associated with optic atrophy, microphthalmia, agenesis of the corpus callosum, and Dandy-Walker malformation. The risk of significant abnormalities is low and has been estimated at 6.4% (95% confidence interval 4.6–8.9%).172 Warfarin exposure is also associated with an abnormally high incidence of spontaneous abortion and intrauterine fetal death.173 The disadvantages to the use of warfarin in pregnancy are such that it should be reserved for the rare situation in which it is impossible to use heparin or dicumarol.174
Antithyroid medications
The fetal thyroid begins to function around the fourth month of gestation. At any time thereafter, antithyroid drugs, such as the thioureas, can inhibit fetal thyroxin synthesis and produce a congenital goiter. These goiters are not large, regress spontaneously after birth, and are rarely associated with hypothyroidism. Goiter has been observed in about 10% of mothers so treated, and the incidence of goiter is not dose-related. It has also been observed in one of a pair of twins, thus emphasizing the capricious incidence.175
The goal of the treatment of mothers with Grave's disease should be aimed at keeping maternal free thyroxine concentrations close to the upper limits of normal for non-pregnant patients.176 The use of iodine-125-labeled fibrinogen, frequently used for the identification of deep venous thrombosis, is contraindicated in pregnancy because radioactive iodine crosses the placenta and is concentrated by the fetal thyroid after the 12th week of pregnancy. Povidone–iodine or iodine-containing solutions used for disinfection during labor and delivery can be absorbed and transiently affect thyroid function in the newborn.177 Propylthiouracil and methimazole are used for treatment of maternal hyperthyroidism; of the two, the former appears to be clinically preferable, because it may be transferred into the fetal circulation less readily than is the latter.178 Also, methimazole has been associated with scalp defects known as aplasia cutis.179
Hypothyroidism during pregnancy
There is compeling evidence indicating that children of mothers with hypothyroidism untreated during the first trimester of pregnancy, may have diminished intellectual cognitive abilities. Treatment with levothyroxine of women with subclinical hypothyroidism and positive thyroid antibodies remains controversial.180
Adrenal steroid hormones
Prednisone and related compounds are not uncommonly used during pregnancy for the treatment of a wide variety of disorders. Concern over possible effects on the fetal pituitary–adrenal axis has been tempered by the knowledge of the poor placental transfer of prednisone. Although the overwhelming majority of infants born to mothers receiving prednisone have normal cortisol production, several cases of hypoglycemia with or without hypoadrenalism have been reported. Spontaneous recovery is almost always universal. The rare reports of neonatal deaths due to adrenal insufficiency proven by postmortem examination occurred in infants whose mothers received high corticosteroid doses.
Anti-infective drugs
Several studies have indicated that the pharmacokinetics of beta-lactam antibiotics is altered during pregnancy.181 The elimination of ampicillin is increased during pregnancy. In one study the maximal and minimal concentrations of ampicillin at 10 weeks of gestation and at term after an equivalent dose were found to be less than two-thirds and approximately half the concentrations, respectively, of those obtained in nonpregnant women.182 Cefazolin, cefuroxime, and cephradine have significantly decreased plasma concentrations and half-lives during pregnancy compared with those obtained in the nonpregnant state.183, 184
Most antibiotics cross the placenta even some with molecular weights higher than 1000 kDa (e.g., vancomycin and colistimethate). A handful of antibiotics (e.g., ampicillin, methicillin) cross the placenta rapidly with equilibration of fetal and maternal concentrations. Most antibiotics are transferred incompletely. This group includes azlocillin, dicloxacilin, pipercillin, amikacin, gentamicin, kanamycin, streptomycin, fosfomycin, griseovulvin, vancomycin, and colistimethate.185 Molecular weight is an important but not exclusive determinant of the extent of placenta transfer. Other factors including maternal protein binding and specific transport process modify the rate of transfer across the placenta.
As HIV infection becomes more prevalent in women, the use of antiretroviral drugs during pregnancy requires special consideration for the safety of the fetus.
There is ample evidence that combined antiretroviral therapy (ART) can significantly reduce the transmission of human immunodeficiency (HIV) to less than 2%.186 There is a paucity of information concerning the phamacokinetics data on antiretroviral therapy given during pregnancy.187 Zidovudine pharmacokinetics has been studied in a limited number of pregnant women. No difference was found in total body clearance and half-life between pregnant and nonpregnant patients.188 There are data on the pharmacokinetics of lamivudine, didanosine, and stavudin. There are no significant changes in the Pk of these drugs in relation to non-pregnant women. The role of concentration-targeted ART has not been investigated. Pharmacodynamics effects of ART (e.g., measurements of viral loads, T cell counts) are used in the management of mothers treated with ART.
It is likely that the antiretroviral biodisposition for renally eliminated drugs such as lamivudine and indinavir, and highly protein-bound drugs such as protease inhibitors may be affected by pregnancy. Unfortunately, the accelerated approval of new antiretroviral agents does not include pharmacokinetics and long-term safety studies in pregnancy.
Zidovudine pharmacokinetics has been studied in a limited number of pregnant women. No difference was found in total body clearance and half-life between pregnant and nonpregnant patients.189 Concern has been expressed regarding the risk of mitochondrial toxicity associated with the use of antiretroviral nucleoside analogues during pregnancy.190
DRUGS GIVEN DURING LABOR AND DELIVERY
Analgesics
Although systemic analgesia has been used in obstetrics for decades, concerns about the occurrence of maternal and fetal side effects has led to the use of epidural anesthesia during the early stages of labor. Systemic analgesia is widely used as an alternative when epidural anesthesia is not feasible.
A number of opioids and procedures of administration are being use but definitive large control trials for safety and efficacy are not available. Meperidine is one of the analgesics most widely used during the first stage of labor. After intramuscular administration, peak concentrations are obtained 20–30 minutes later. The absorption rate varies with the injection site and regional blood flow, and appears to be slower in women during labor than in healthy volunteers.191 With repeated intramuscular injections, marked variations in blood concentration result in unpredictable pain relief.192 The volume of distribution is significantly reduced when compared with controls. Meperidine is extensively metabolized to normeperidine and several other derivatives.
Biodegradation is the major pathway of meperidine elimination, because less than 5% is excreted unchanged in the urine. Pharmacologically, normeperidine retains about half the analgesic activity of the parent compound. The striking interpatient variability in body clearance and distribution may help explain differences in clinical response.
The standard clinical practice of using single or multiple doses of 50 mg irrespective of the patient's weight is partially responsible for blood concentrations that may be two to four times higher than needed for analgesia. These concentrations are well into the toxic range.
The following factors appear to be important in determining the amount of meperidine and normeperidine transferred to the fetus: dosage, route of administration, dose-delivery intervals, and maternal metabolic rate. Stress or relative hypoxia may induce hemodynamic changes (e.g., decreased liver and renal blood flow) that may be responsible for reduced meperidine disposition in both mother and newborn.193 There is considerable evidence supporting the view that normeperidine is more toxic than meperidine and is only in part antagonized by naloxone. The demonstration that previous or concomitant phenobarbital treatment induces meperidine biodegradation, thereby increasing the amount of its toxic metabolite normeperidine, is of considerable clinical significance.194
Significant concentrations of normeperidine have been found in blood samples obtained at birth or immediately thereafter, lending support to the theory that this toxic metabolite plays a pivotal role in neonatal meperidine intoxication. The apparent lack of correlation between central nervous system depression and meperidine blood concentrations at birth corroborates this postulate. To the well-known alterations in meperidine disposition during pregnancy, one must now add profound kinetic changes in women during labor.
The use of naloxone for the prevention of analgesic-induced respiratory depression in the newborn is not recommended for administration to the mother before delivery because it can reverse maternal analgesia. In addition to its questionable efficacy under these circumstances, naloxone, by acting on receptors on the spinal cord, brain stem, pituitary, and thalamus, can alter the release of endogenous endorphins. Evidence of the protective role of endorphins against fetal stress and asphyxia is mounting.195, 196 Meperidine and butorphanol (Stadol) are known to produce a benign type of sinusoidal fetal heart rate pattern.197 This pattern is usually considered a sign of serious fetal distress. Butorphanol, in contrast to meperidine, is metabolized to an inactive metabolite. Other opioids used for obstetric analgesia includes morphine, fentanyl, alfentanil, sufentanil, and remifental.
Morphine is a powerful depressant of the fetal nervous system (10 times more depressant than meperidine). Fentanyl offers the versatility of different routes of administration (i.e., oral, transdermal patch, subcutaneous). A dose-dependent adverse effect on infant feeding has been described but not documented by other studies. Remifentanil, a μ receptor agonist appears to be less toxic than other opioids, with fast initiation of action and predictable pharmacokinetics. In recent years, there has been an increase in the use of patient controlled intravenous analgesia. Using this delivery method prolonged analgesic effect may ensue with pain relief extended to subsequent contractions.198 The role of remifentanyl in obstetric analgesia needs to be substantiated by large controlled clinical trials.
Regional anesthetics
Because of its effectiveness and relative safety, regional anesthesia for labor and vaginal delivery has gained wide acceptance. This type of conduction anesthesia includes spinal, lumbar epidural, caudal, paracervical, pudendal, and local perineal infiltration. Although there are a bewildering number of local anesthetics available, modern regional anesthesia is practiced with a selected number of compounds. Local anesthetics are divided into two main pharmacologic groups, depending on their molecular configuration: ester linkage (e.g., procaine) and amide linkage (e.g., lidocaine). The fetal and neonatal toxicity of local anesthetics depends on the amount of free (i.e., nonionized and nonprotein-bound) drug that reaches the fetus. Many factors determine the amount and rate of free drug entering the fetal compartment. They are total dose, route of administration, presence of vasoconstrictive agents in the anesthetic solution, rate of maternal metabolism and excretion, maternal and fetal pH, pKa of the drug, and degree of maternal and fetal protein binding.
The amount of anesthetic used in spinal anesthesia is too small to reach significant fetal levels. Absorption from other injection sites depends on local vascularity and the presence of tissues such as fat capable of binding local anesthetics. Vasoconstrictor drugs such as epinephrine, octapressin, and phenylephrine are often added to antagonize increases in local blood flow secondary to vasomotor blockage and to retard absorption of local anesthetics. Generally, epinephrine reduces peak plasma concentrations of local anesthetics in most of the commonly used regional blocks. The effect of vasoconstrictors is most marked with lidocaine, in which case the duration of the anesthetic effect may be prolonged by as much as 50%. Although epidural dose requirements of local anesthetics are reduced in pregnancy because of engorgement of vertebral veins, this is not associated with changes in their absorption rate. Decreased metabolism is probably a more important contributing factor.
The amide type of anesthetic, namely bupivacaine, lignocaine, and mepivacaine, is metabolized by hepatic amidases. In nonpregnant subjects, the elimination half-lives for the commonly used local anesthetics range from 1.5 hours for lidocaine to 3.5 hours for bupivacaine.199 Epidural administration of bupivacaine for cesarean section results in a biphasic maternal blood elimination rate: an initial phase with a half-life of 47 minutes and a slow elimination phase with a half-life of 9 hours.200 Further kinetic studies are needed, especially during normal and abnormal labors.
In contrast to the amides, the esters (tetracaine, chloroprocaine) are biodegraded by both hepatic and plasma esterases. Hydrolysis by plasma pseudocholinesterase is quite fast (less than 1 minute for chloroprocaine), a welcome feature because it reduces the risk of fetal intoxication. No difference in the rate of biodegradation of 2-chloroprocaine has been found between preterm and term fetuses.201 The major drawback of ester-linked agents is the short duration of their pharmacologic effect.
Fetal uptake and distribution of local anesthetics are influenced by several factors. Although it has been postulated that the high protein binding of certain local anesthetics (e.g., 95% for bupivacaine) would affect placental transfer, experimental data do not support this contention. The significantly lower fetal plasma binding of local anesthetics, however, may have a profound effect on fetal distribution. Indeed, when compared with bupivacaine, the greater neurobehavioral disturbances seen after maternal administration of lidocaine seem to be related to the higher concentration of free drug in the fetus.202 Fetal acidosis may facilitate placental transfer of local anesthetics by the phenomenon of “ion trapping”. Human and experimental data support this contention.203 Because of the rapid maternal biodegradation of 2-chloroprocaine, placental transfer is not increased even in the presence of acidosis.
Clinical manifestations of local anesthetic intoxication in newborns include bradycardia, apnea, hypotonia, mydriasis, and, on occasion, seizures. Animal studies have demonstrated that bradycardia is due to the toxic effect of local anesthetics on the myocardium. Seizures have been mostly associated with accidental mepivacaine overdose, usually occurring during the first 6 hours of life. In addition to the obvious clinical picture of neonatal cardiovascular and central nervous system depression, that sometimes follows obstetric regional anesthesia, some studies have focused on more subtle and long-lasting neurobehavioral disturbances.204 Lidocaine affects the neurobehavior of the neonate to a greater extent than chloroprocaine.205 The fetus may be affected not only by the direct action of local anesthetics but also indirectly by changes effected by these drugs on the maternal organism. Areas in need of further investigation include the maternal and fetal disposition of local anesthetics during labor and in high-risk pregnancies, interaction with other compounds, and the effect of metabolic and acid–base disturbances on the distribution of the different compounds.
General anesthetics
Inhalation anesthetics diffuse rapidly and easily across the placenta. Two groups can be distinguished: those such as nitrous oxide that are excreted by alveolar ventilation without prior metabolism, and compounds such as trichloroethylene and methoxyflurane that undergo different degrees of metabolic degradation.
The general anesthesia technique requires maternal denitrogenation and induction before intubation with either sodium thiopental or ketamine. Although the use of ketamine may be associated with fewer neurobehavioral effects in the neonate, maternal side effects (i.e., hallucinations) limit its use. Before induction and during surgery, small doses of muscle relaxants are often used. Nondepolarizing agents (including the newer short-acting vecuronium and atracurium) or succinylcholine in usual doses do not cross the placenta in significant amounts. The concurrent use of magnesium sulfate potentiates the effect of nondepolarizing agents during general anesthesia.
Nitrous oxide has been used as both an analgesic and an anesthetic. Experimentally, preinduction with thiopental followed by 70% nitrous oxide has produced fetal acidosis. At concentrations of 75%, which are required to produce anesthesia, significant depression at birth occurs in a sizable number of infants. Secondary apnea has been attributed to alveolar diffusion of the gas and may lead to dilutional hypoxemia. Current anesthesia practice is to supplement 50% nitrous oxide with low concentrations of potent inhalation agents (e.g., halothane, isoflurane, enflurane) to prevent maternal recall and awareness and thus reduce the high catecholamine concentrations associated with light anesthesia. In general, inhaled anesthetics will not depress the fetus if the induction to the delivery time is not prolonged. Higher concentrations of potent inhalation agents, by decreasing maternal cardiac output and blood pressure, may reduce uterine blood flow and result in fetal acidosis.
A potential source of concern has been the inhibitory effect of nitrous oxide on methionine synthesis.206 Because of the short fetal exposure, this fact is unlikely to be clinically significant. Beside fetal depression, inhalation agents are not known to injure the fetus in ways that are obvious at present.
DRUGS GIVEN TO STOP PREMATURE LABOR
Premature labor is a complex and multifactorial disorder; uterine contractions may constitute only a symptom. It remains as a formidable challenge for obstetricians in the next decade. The lack of uniform success with any of the available tocolytic agents underscores the lack of specificity of the available therapy.207, 208
Ritodrine hydrochloride is the only FDA-approved tocolytic agent. Because of infrequently reported but severe treatment-related complications, there is currently a trend away from the use of beta-sympathomimetics to other drugs or even combination therapy.
Beta-adrenergic stimulants
Several beta-sympathomimetic drugs have been used for inhibition of preterm labor. The list, in addition to ritodrine, includes salbutamol, fenoterol, isoxsuprine, and terbutaline. Ritodrine crosses the placenta and reaches concentrations in fetal blood approaching those of the mother.209 Complications of ritodrine therapy have been reported after intravenous administration. A recent study found a decreased bioavailability of oral ritodrine.210 Maternal complications include cardiovascular and renal dysfunction and metabolic disturbances. Pulmonary edema remains the most serious maternal complication. Pregnant women at risk for pulmonary edema are those receiving concomitant glucocorticoids and those with preeclampsia or twin pregnancy. The etiology of pulmonary edema remains elusive, but fluid volume overload seems to play a contributory role. Ritodrine is known to reduce renal blood flow and creatinine clearance; renal insufficiency, however, is an unusual complication. Hypotension, not uncommon with isoxsuprine therapy, does not occur with ritodrine infusion with the exception of bleeding patients (i.e., abruptio placentae or placenta previa).
Metabolic alterations associated with the use of beta-adrenergic agonists include mild hyperglycemia and increased concentrations of lactate and free fatty acids. Long-term infusion of ritodrine to pregnant women has not been associated with significant changes in metabolic parameters. A single report, however, describes fasting hyperinsulinemic hypoglycemia after prolonged ritodrine therapy in a pregnant woman.211 The short-term effects of ritodrine in the fetus mirror those in the mother. Drug-related side effects in the newborn are less pronounced than those associated with the use of isoxsuprine. Hypotension, abdominal distention, and paralytic ileus, previously reported complications of maternal isoxsuprine use, have not been found in more recent studies of the neonatal complications of intrauterine exposure to ritodrine.212 Neonatal hypoglycemia and renal dysfunction are complications of ritodrine therapy the true incidence of which remains unknown; this area requires further study.213 A transient leukemoid reaction in both mother and newborn has been associated with maternal ritodrine therapy.214
The use of magnesium sulfate, as a tocolytic agent, used either alone or as part of combination therapy (i.e., with ritodrine) has diminished in recent years. A large placebo controlled trial failed to demonstrate efficacy. Neonatal hypotonia, the most common side effect, does not correlate with peak plasma levels. The elimination half-life at birth is approximately 40 hours; consequently, normal magnesium plasma levels may not be reached until 1 week of age. Interest in the use of magnesium sulfate has shifted from its role as a tocolytic drug to that of a fetal neuroprotective agent. There is both epidemiological and basic science data supporting this application. Recently an analysis of four trials that enrolled over 3700 patients was undertaken.215 No significant effects in major clinical outcomes including mortality and neurological outcome in the first year of life were detected. There was, however, a marked decrease in significant gross motor dysfunction in the magnesium treated group. Follow up studies in these and other trials are needed to ascertain the significance of the reported findings.
Indomethacin is another tocolytic agent regaining popularity. Controlled European studies rebutted previous anecdotal reports of pulmonary hypertension and fetal death.216 Concerns about fetal bleeding and other potential hazards limit the use of indomethacin to research studies only.
The use of calcium antagonists (i.e., nifedipine) to suppress preterm labor should be considered investigational, and claims of their superiority to ritodrine remain contentious.217
Oxytocin receptor antagonists have been recently tested but there is no convincing evidence that they substantially reduced the incidence of premature birth. The use of progestins to prevent premature birth in women with other risk factors is currently being investigated.218 In a large multicenter trial conducted by the Maternal-Fetal Medicine Unit Network using weekly intramuscular injections of 17-alpha hydroxyprogesterone caproate (17-OHPC) a decrease in the rate of preterm birth (33%) was observed.219 The mechanism by which 17-OHPC reduces the preterm birth remains elusive. A recent study ruled out plausible mechanisms such as a greater affinity for progesterone receptors or glucocorticoid receptors of a facilitated stimulation of progesterone responsive genes compared to progesterone.220
DRUGS GIVEN TO INDUCE LABOR
Oxytocin continues to be the drug of choice for inducing labor. Careful monitoring of fetal heart rate, uterine action, and accurate dosing to avoid sudden changes in blood levels, as well as clear delineation of its contraindications, have minimized fetal side effects. There remains some controversy over the range of starting dose and time interval before the dose is increased. There is also disagreement as to the desired response, with some authors considering progressive cervical dilatation the appropriate response, whereas others221 suggest that it is uterine activity. Several studies have been conducted with the aim of optimizing oxytocin treatment and minimizing fetal side effects. It was found that increasing the duration of infusion before stepping up the dose rate might decrease the delivery time.222 This finding appears related to the time required to reach a steady state (i.e., a premature increase in the dose may result in maternal or fetal toxicity). Due to the molecular size of oxytocin and the presence of oxytocinase in the placenta, it is considered unlikely for oxytocin to cross the placenta in significant amounts. There is epidemiologic evidence linking the use of oxytocin with significant neonatal jaundice; the nature of this association remains obscure. Controversy exists as to whether oxytocin or induction per se is responsible. There is evidence that fetal immaturity is the responsible factor.223
The use of prostaglandin E2 (PGE2) as an adjunct to oxytocin has increased, with the goal of reducing the high number of failed induction rates. Although PGE2 has been found effective by several authors, the use of prostaglandins alone is probably not sufficient in all instances. Typically prostaglandins are used to soften the cervix and change receptors, and then oxytocin is used because its control of uterine contractions is more precise.224 PGE2 should not be continued or administered in active labor because it may lead to hyperstimulation. Several studies have attempted to determine which prostaglandins are more effective at cervical ripening. The type of PGE2 preparation as well as the delivery system and route of administration influence the results. The use of dinosprostone low-release vaginal insert was found to be effective for elective induction of labor in postdate pregnancy.225
ANTEPARTUM GLUCOCORTICOID ADMINISTRATION AND PREVENTION OF HYALINE MEMBRANE DISEASE
A vast amount of literature exists on the use of glucocorticoids to prevent hyaline membrane disease, or respiratory distress syndrome.226 Most clinical trials involve the administration of either betamethasone or dexamethasone. It is generally agreed that prenatal steroid administration is effective in decreasing the overall incidence of respiratory distress syndrome. Analysis of the combined results of six randomized double blind trials revealed an incidence of respiratory distress syndrome in 29% of the placebo group and 8.5% of the steroid-treated group (a highly significant difference).227 A marked effect of sex and race has been noted, with blacks and female infants more likely to benefit from steroid therapy. In 1994, the National Institutes of Health sponsored a Consensus Development Conference to assess the effectiveness of antenatal steroids. They concluded that a single course of corticosteroids given to women at risk for preterm delivery reduces the risk of death, respiratory distress syndrome, and intraventricular hemorrhage in their offspring.
In recent years, the practice of using repeat courses of antenatal corticosteroids became widespread. This practice fostered vigorous controversies regarding the need, indications, effectiveness, and safety of this approach. In 2000, the National Institutes of Health convened another Consensus Development Conference to review the available evidence on the risks and benefits of repeat courses of antenatal corticosteroids. It was noted that animal studies clearly substantiated deleterious effects on lung growth, retinal development, cerebral myelinization, and function of the pituitary–adrenal axis. The consensus panel concluded that available data are inadequate and that repeat courses “should not be used routinely” unless patients are enrolled in randomized controlled trials.227
A recent international multicenter randomized trial was undertaken in women at risk of preterm birth and who had received a single course of antenatal corticosteroids, the women were allocated to receive multiple courses of steroids or placebo. The study failed to show improvements in outcome in the treated group. Growth, however, was decreased with significant reductions in birth weight, head circumference, length, and head circumference.228 No beneficial effect of prenatal steroids has been demonstrated in twin pregnancies, however, this may be a consequence of inadequate dosing.
Controversy surrounds the use of corticosteroid therapy in cases of preterm premature rupture of membranes. A recent survey of maternal–fetal medicine specialists failed to show consensus in this area. Although lingering doubts remain as to the safety of antenatal steroids, risks in humans seem low and potential long-term effects remain hypothetical. Whereas the ideal candidates for antenatal glucocorticoid administration constitute a small proportion of the overall population at risk for preterm delivery, the benefits to be derived from lung maturation in these fetuses outweighs potential risks.229, 230 The use of other substances to prevent respiratory distress syndrome (i.e., intra-amniotic thyroxin or maternally administered TSH) remains investigational, requiring randomized controlled clinical trials.
SOCIAL HABITS AND LIFESTYLE: THEIR INFLUENCE ON THE FETUS
Cigarette smoking and ingestion of alcoholic beverages and caffeine (coffee, tea, cola drinks, chocolate bars) are habits so ingrained in our culture that we would be hard-pressed to name a handful of our acquaintances who totally abstain from all of them. The evidence incriminating heavy drinking and smoking as intoxicants (noxious) for the fetus seems incontrovertible. The problem facing the obstetrician is to translate the voluminous clinical epidemiologic and animal experimental data into clinical and public health practice. From the outset, it should be clear that the extreme abuse of these social habits could seriously damage the fetus. Unfortunately, the evidence against lesser degrees of abuse is not available as yet (and this affects the majority of patients). A crude dose–response relationship has been reported for alcohol exposure during pregnancy. Drinking behavior in the month preceding the recognition of pregnancy strongly correlates with the occurrence of the dysmorphic abnormalities that have been termed fetal alcohol syndrome. The syndrome also includes varying degrees of prenatal and postnatal growth retardation, and mental deficiency.
Patients with fetal alcohol syndrome have been found to have normal intelligence but have shown evidence of behavioral and learning disabilities.231 Patients exhibiting mild dysmorphic features have also been born to mothers who drank heavily during pregnancy. The lower limit on heavy drinking has been placed as five or six drinks on one occasion, with at least 45 drinks per month.232 The danger from light drinking (less than 1 oz of absolute alcohol daily) has not been demonstrated. It has been suggested that 100 g of ethanol around the time of ovulation can be detrimental to subsequent fetal development.233 A recent study found no apparent ill effects in offspring of women consuming 61–66 g per ovulation week.234 In the same study, alcohol intake of 600 g during the first 12 weeks of gestation (an average of three or four drinks weekly) did not affect organogenesis. An important caveat to be considered in evaluating these findings is that many of the fetal alcohol effects (i.e., behavioral and learning disabilities) are not detected until later on in childhood. In recent years, the widespread publicity about fetal alcohol syndrome has resulted in a sharp decline in alcohol consumption after the detection of pregnancy.235 Undue anxiety and even neurotic reactions have occurred, however, among women who have consumed some alcoholic beverage around the time of conception. Compilation of data from both the United States and the United Kingdom indicates that one or two drinks weekly during this period does not affect fetal outcome. Abstinence during pregnancy, however, remains the foolproof guarantee against fetal alcohol effects. There is a need to identify women who continue to drink while pregnant. A number of biomarkers of alcohol abuse during pregnancy have been identied but there are no validated studies to ascertain their usefulness.236
There is plentiful evidence that cigarette smoking adversely affects the course of pregnancy. It is not known which component of tobacco smoke is responsible for the increased fetal morbidity. Not frequently appreciated is the fact that smoking results in the absorption of thousands of different compounds, including carcinogenic and mutagenic substances. Information concerning the fetal blood concentration of the three major toxins (nicotine, CO, and thiocyanate) is sparse and scattered throughout the medical literature.237, 238 One recent study found that the fetus of a smoking mother is exposed to higher nicotine plasma concentrations than is the mother. Although the complex composition of cigarette smoke makes it difficult to relate pharmacologic and toxic effects to a single compound, there is a significant body of evidence linking nicotine to a wide range of pharmacologic and physiologic effects on the fetus.239, 240
Marijuana is another social drug in our culture, regardless of its legal status. Whereas the teratogenic effects of cannabis and 9-tetrahydrocannabinol are firmly established, 112 clinical studies have failed to provide proof of fetal toxicity.241 Abnormalities in the duration of labor, lower birth weight, shorter gestational age, and increase in the incidence of major malformations among marijuana users have been reported by different investigators.242, 243 A single report describes a constellation of anomalies resembling those found in the fetal alcohol syndrome in five infants born to heavy users.244 Other studies have failed to find fetal abnormalities in the offspring of moderate to heavy marijuana users. Small sample sizes, short length of follow-up, and low relative risks can explain negative results. The belated recognition of the relationship between alcohol and cigarette smoking should serve as a warning and stimulate larger epidemiologic studies.
In addition to marijuana, the fetus may be exposed to a whole array of addictive substances, including narcotics, amphetamines, chlordiazepoxide, phencyclidine (PCP), and others. Intrauterine exposure to heroin, methadone, or heroin substitutes (“Ts and blues”, pentazocine, and tripelennamine) can produce at birth similar withdrawal symptomatology at birth.245
The recreational use of cocaine, alone or in combination with methamphetamine (“crystal”), has increased dramatically in recent years. The burden of amphetamine abuse has also been a major concern in recent years particularly in younger women and those living in rural areas.246 Increased rates of prematurity, intrauterine growth retardation, and placental hemorrhage have been reported as a consequence of in utero exposure to these two drugs.247 Both cocaine and methamphetamine act as sympathomimetic compounds, and thus it is likely that some of the reported complications (i.e., abruptio placentae, perinatal cerebral infarction)248 are due to vasoconstriction. Cocaine is metabolized by cholinesterases, which are known to be low in the fetus, therefore enhancing prolonged toxic exposure. One study found a high malformation rate in the offspring of pregnant women abusing cocaine exclusively.249 The teratogenesis of cocaine remains contentious; further large-scale epidemiologic studies are required.
The clinical manifestations of neonatal withdrawal from most abused drugs are part of a continuum between normal and aberrant behavior with or without abnormal somatic development and autonomic symptomatology. Rarely are the symptoms drug-specific (i.e., sudden outburst of agitation, flapping tremors with sudden changes in behavior in PCP-exposed infants).250 Because multidrug abusers predominate, it is difficult to isolate the effects of a single drug.
Unquestionably, the most widely used of the “social drugs” is caffeine. The charge against caffeine is mostly indirect and stems from animal and epidemiologic clinical studies. Documentation of harmful effects on the fetus is elusive because of its universal use, making it difficult to obtain a population not exposed to it, and because of the strong association between heavy caffeine intake and alcohol and smoking habits.
Although it is clear that additive drugs and chemicals should be prohibited during pregnancy, a more flexible attitude should be adopted with the various social habits. Better compliance and less anxiety would be generated if physicians would advise their patients of the relative risks of the various social habits.
The use of herbal medicines by pregnant women has increased dramatically in recent years. It has been estimated that almost a third of pregnant women use complementary medicines and therapies without the knowledge and consent of obstetricians.251 The herb blue cohosh, considered a human fetal toxin, has been used to induce labor.252 Two reports describe toxic reactions in infants at birth. One newborn was born with congestive heart failure secondary to myocardial infarction;253 another newborn presented with seizures due to encephalopathy and renal failure.254 Other herbs used in pregnancy are potentially harmful to the fetus. For example, berberine, the major ingredient of the Chinese herb huanglian, can be associated with significant jaundice in the newborn. Berberine also displaces bilirubin from albumin, thus increasing the risk of kernicterus.255 Herbal medicines (e.g., Saint John's wort) may significantly alter the biodisposition of a number of drugs (e.g., digoxin, carbamazepine, and protease inhibitors).
FETAL THERAPY: A HOPE FOR THE FUTURE
It is likely that in the future, medical and pharmacologic manipulations of the fetus may overshadow surgical interventions as the principal forms of fetal therapy.
Transplacental therapy for the control of fetal tachyarrhythmias is a well-accepted treatment option. Unfortunately, different drugs have been used in an uncontrolled fashion. Digoxin, flecainide, sotalol, and verapamil have been variously used as the initial treatment. Digoxin is the most commonly used drug for this purpose, although a higher success rate was reported for flecainide.256 Except for digoxin, no pharmacokinetic data are available for antiarrhythmic drugs in pregnancy. When the initial treatment fails, combination therapy is used. The demonstration of the existence of P-glycoprotein in the mouse placenta raises the possibility of drug–drug interactions. It is known that verapamil inhibits the excretion of digoxin by inhibiting its transport by P-glycoprotein. Under these circumstances, high concentrations of fetal digoxin may occur.257
The successful pharmacologic suppression of the fetal adrenal gland by maternal administration of dexamethasone to prevent masculinization of the fetus in cases of 21-hydroxylase deficiency illustrates an attempt at pharmacologic fetal therapy.258
Vitamin therapy has been attempted for the fetal treatment of vitamin-responsive types of genetic metabolic disorders (i.e., vitamin B12 for methylmalonic acidemia, biotin for multiple carboxylase deficiency). At this time, it is not possible to ascertain the effectiveness of these and other types of fetal interventions. Direct instillation of substances into the amniotic fluid or intestinal gut (i.e., enzymes to prevent meconium ileus in cystic fibrosis) is a therapeutic modality that may be exploited in the future.
OBSTETRIC AND FETAL PHARMACOLOGY
The development of obstetric and fetal pharmacology as a new discipline is contingent on the removal of obstacles to the widespread study of drugs in pregnancy. Some of these problems and possible solutions are summarized below.
Off-label prescribing
Drugs used during pregnancy are prescribed for obstetric or medical conditions. Several drugs used for obstetric indications have been labeled for other indications in nonpregnant adults (e.g., nifedipine and indomethacin as tocolytic agents rather than antihypertensive and anti-inflammatory drugs, respectively). Off-label prescribing refers to the practice of prescribing drugs for indications other than those listed on the product label. Because of the lack of obstetric testing and labeling, off-label prescribing is the norm in obstetrics.259
Physicians must prescribe drugs based on acceptable evidence. Unfortunately, in obstetrics the information available is not based on sound scientific evidence. Physicians must rely on published experience consisting of case reports, small trials, and aggregation of studies close to the end of pregnancy. Many studies are retrospective, and there is a dearth of dosing and bioavailability studies. The trial-and-error approach to the use of drugs in pregnancy has led to unfounded generalizations and untested assumptions. Because of the lack of evidence, each patient becomes an experiment. Drugs are just “tried out” without knowledge as to whether they will work, and decisions about doses are often educated guesses.
The Physician's Desk Reference is the most widely used source of drug information for physicians. The most common statement in this reference for most of the drugs used in obstetrics is: “The safety and effectiveness of this drug has not been established in pregnancy.” Some drug labels go even further; the manufacturer's label of terbutaline states categorically, “Terbutaline sulfate should not be used for tocolysis.”
Drug development process: labeling of drugs
The FDA must approve all indications listed on a drug product label or package insert. The approval process ensures that the FDA requirements for safety and efficacy are met.
The clinical trials included in the drug development process comprise four phases. Phase I is mostly concerned with safety and is generally done in healthy volunteers. Phase II includes controlled trials for therapeutic efficacy in well-matched groups; dose regimen pharmacokinetics and metabolism studies are also performed in this phase. Phase III involves large efficacy trials enrolling from several hundred to several thousand patients. A new drug is tested in an average of 64 clinical trials; up to 50% of these trials fail to show conclusive results. After completion of phase III, a voluminous all-inclusive dossier (5000–100,000 pages) that contains both preclinical and clinical data is analyzed by an FDA team of reviewers to determine whether the drug is safe and effective. This process applies to drugs with specific obstetric indications (e.g., a new tocolytic agent with no other adult indications).
Most drugs used in obstetrics have been labeled for nonpregnant adults. Under these circumstances, pregnant women constitute a special population. The requirements for labeling of drugs in special populations vary according to the drug type, indication, and characteristics of the disease process in the special population.
The process of labeling drugs with adult indications in pediatrics is a good model that is applicable to obstetrics. Safety and pharmacokinetic or pk-pd studies are required. In 1994, the FDA established the Final Pediatric Rule. This rule, which applies to approved drugs and biologicals, states that extrapolation of adult efficacy data would be permitted if the FDA concludes that the course of the disease and the effects of the drug, both beneficial and detrimental, are sufficiently similar in children and adults. This approach could be applied to obstetrics.
Pharmaceutical industry and study of drugs in pregnancy
The FDA does not require the study of drugs in pregnancy. To effect a change in label (e.g., use of a drug in pregnancy), a pharmaceutical company must file a supplemental application and perform studies for the specific indication in pregnancy as negotiated with the FDA. Pharmaceutical companies, because of fears of fetal harm and financial loss resulting from liability claims, have been reluctant to study drugs in pregnancy. Other important economic considerations include projected sales, time remaining of patent exclusivity, and delay in the time of FDA approval. The lack of incentives to file a supplemental application for obstetrics indications is a major obstacle.
A viable solution would be the creation of economic incentives. The lack of pediatric drug testing and labeling led to the enactment of the pediatric exclusivity provisions of the FDA Modernization Act of 1997. This regulation was designed to encourage pediatric labeling. The FDA issued new regulations in August 1997 (Final Rule of 1997), requiring pediatric studies of certain new drugs. In addition, in 1997 the FDA Modernization Act (FDAMA) was enacted. This legislation contained a provision (Section 111) that provided exclusivity incentives for the pharmaceutical industry (6 months of additional patent exclusivity) to conduct pharmaceutical trials in children. The pediatric provisions of FDAMA expired in December 2001. The Best Pharmaceuticals Act for Children (BPCA), enacted in January 2002, extended the exclusivity incentives for 5 years. The BPCA also provides a mechanism for the study of off-patent drugs and identifies the need to conduct studies in the newborn population. The 2007 reauthorization of the BPCA and the Pediatric Research Equity Act (PREA) increased FDA’s authority to require studies. These acts strengthened adverse event surveillance, and improved the transparency, oversight, and administration. Legislative and regulatory remedies have been very successful in encouraging pharmaceutical companies to study drugs in children. Economic incentives and more liberal regulatory requirements could lead to "deorphanize" the obstetric population.
Legal issues
The overwhelming concern of pharmaceutical companies is the possibility of liability claims for fetal harm as a consequence of fetal drug exposure. It could be argued that if drugs are widely used, not obtaining evidence constitutes willful negligence. However, pharmaceutical manufacturers rely on the “learned intermediary” doctrine. The industry's contention is that they manufacture drugs, but physicians prescribe them. Nevertheless, if there is a negligent failure to obtain data in the first place, the physician cannot “learn” in the absence of data. Actually, neither the physician nor the manufacturer should be liable. There is a need to modify the tort liability system applicable to drug studies in pregnancy.
Ethical issues
Ethical issues related to the study of drugs in pregnancy are not dissimilar to those faced by investigators performing drug studies in children.260, 261 The experience in pediatrics can be applied to the use of pregnant women as subjects of drug research. In 1997, the American Academy of Pediatrics made the pronouncement that it is unethical not to study drugs in children. Similarly, there is a moral imperative to advance the therapeutic management of pregnant women.262 The benefits of research must be available to all segments of the population, regardless of gender, race, ethnicity, age, and subpopulations such as pregnant women. There are, however, ethical concerns for studies performed during pregnancy. Ethical guidelines for such studies need to be developed. Placebo or control trials can be ethically justified only for the treatment of serious obstetric conditions for which no therapy is currently available. Phase I clinical trials must not include pregnant women. The issue of fetal therapeutics is more complex and contentious. There is an inherent conflict of interest when the mother is used as a vehicle to treat the fetus. Ethical guidelines for fetal therapy must be established.
Obstetric Pharmacology Research Unit (OPRU) Network: proof of concept
The NICHD had a dual purpose in creating the OPRU Network in 2004: to prove the concept that drugs that are of therapeutic value during pregnancy can be studied in an ethical fashion and to document the alterations produced by the pregnant state in the biodisposition and effects of drugs in normal or abnormal pregnancies. The goals of the OPRU Network are (1) to serve as a resource for pharmacologic studies of drug disposition and effect during normal and abnormal pregnancies; (2) to conduct single-site and multi-site cooperative phase I and II clinical trials; (3) to conduct pharmacogenetic studies on the effect of pregnancy on drug metabolizing enzymes, transporters, and effectors; (4) to perform studies of placental transfer of drugs; (5) to conduct studies of fetal and maternal pharmacology; (6) to facilitate the utilization of clinical materials for basic research studies; and (7) to enhance the exchange of information between basic scientists and obstetricians and among various specialists involved in treating pregnant women. The OPRU also serves as a resource for the training of health professionals in obstetric-fetal pharmacology and drug trials in pregnant women.263
During the current funding cycle the Network has performed intensive pharmacokinetic and pharmacodynamic studies and adopted an interdisciplinary translation research program including a systems biology analysis. Of note are studies on the effect of pregnancy on drug metabolizing enzymes in different animal models and humans, placental disposition of drugs, and pharmacologic methods development.264, 265, 266, 267, 268
Risk assessment
The teratogenic potential and the fetal adverse effects of different therapeutic drug groups have been detailed in previous sections. Experimental animals have been widely used to establish teratogenic potential, but it is obvious that animal data often have little relevance to the human fetus. Animal models are most useful to unravel the mechanisms of fetal adverse effects.
Description of adverse drug effects in the human fetus is usually in the form of case reports that suggest causation. Additional case reports and retrospective reviews usually follow. Rarely, case–control studies are undertaken. Quantification of risk is often missing, and available information is often misrepresented.269, 270, 271 For example, maternal alcohol intake can produce the fetal alcohol syndrome, but danger from light drinking has not been established. Maternal lithium intake increases the risk of Epstein anomaly from 1 in 20,000 to about 1 in 5000. The risk is small, and the benefits of treating bipolar disorders in pregnancy far outweigh the minimal risk.
Timing of drug intake during pregnancy is of paramount importance in assessing risk. For instance, systemic corticosteroids increase the risk of oral clefts, but exposure after 10 weeks of gestation does not result in oral clefts.
It is generally acknowledged that spontaneous reporting of adverse drug reactions is inaccurate, and underreporting is the rule. Prospective case–control studies and targeted prospective registries should be more widely used. There is a need to coordinate and integrate the different methods used to quantify fetal adverse drug reactions.
A major unresolved problem is the determination of the long-term effects of drugs in the developing central nervous system. The longitudinal cohort study sponsored by the National Institute of Child Health and Human Development and other governmental agencies may be ideally suited to help answer this question. This project would involve about 100,000 children followed from the diagnosis of pregnancy to puberty.
The current efforts by the FDA to update the obstetric risk classification system are encouraging, especially if it contains appropriate information on risk assessment. Pregnant women must be empowered to make informed decisions concerning drug exposure. Most of the time, the risk associated with drugs used in pregnancy is minimal. The widespread acceptance of the concept that there are no risk-free medications will remove a major obstacle to the study of drugs in pregnancy.
Future directions
Intensive pharmacokinetic and pharmacodynamic studies should be conducted for commonly used drugs when there is prima facie evidence of alterations in drug sensitivity or biodisposition. Population pharmacokinetics (i.e., using sparse sampling from patients under routine treatment conditions) can be used to screen drugs in need of further study.
Stable isotope methodology can be used to study pregnancy effects on drug absorption and to evaluate the activity of drug-metabolizing enzymes during pregnancy and postpartum.
During pregnancy, appropriate analytic methods should be used (e.g., high-performance liquid chromatography-mass spectrometry instead of immunochemical methods for digoxin assay). Translational research studies should include estimation of activity of drug-metabolizing enzymes using pharmacologic probes. Phenotypic/genotypic correlation studies are also needed.
The studies in exploratory clinical pharmacology of pregnancy will generate basic research questions. In addition to the study of drug-metabolizing enzymes, the functions of transporters, receptors, and ion channels during pregnancy need to be characterized. Results from these studies will lead to other studies of the genome in pregnancy and fetal and maternal expression of genes involved in the functions of receptors, transporters, and ion channels.
The studies of drug exposure in pregnant women must be complemented by studies of fetal pharmacology. The extent of fetal exposure to drugs given to mothers is largely unknown.
Animal models will need to be used to determine fetal drug exposure and consequent fetal pharmacologic and toxic effects. The combination of fetal pharmacokinetic modeling in appropriate animal models and the use of in vitro systems to study placental drug transport will be needed to estimate fetal drug exposure. The information obtained by this combined approach can then be incorporated in computer simulations and physiologic pharmacokinetic modeling of pregnancy to formulate dosing strategies that could minimize fetal drug exposure.272 Computer modeling would also allow the use of nonsteady-state drug concentrations in mother and fetus at birth to confirm the assumptions of predictive models. Fetal population pharmacokinetics could be used for this purpose.
The application of "omic" technologies has grown in geometric fashion in the post-genomic era, and research studies are beginning to focus in the context of obstetrics.273 Proteomics and metabolomics applications in obstetrics are in the embryonic stage. Once fully developed, these omic research strategies will play a pivotal role in the elucidation of pathophysiology of heterogenous maternal diseases and conditions of pregnancy such as IUGR, preeclampsia, and preterm delivery. Biomarker discovery will lead to the identification of key metabolomic and protein markers that can be used for hypothesis generation and serve as tools in the process of drug discovery. New molecular therapeutic targets will be identified and drugs specifically designed to modulate the desired therapeutic effect will become available.
Alignment of vision between obstetricians, fetal–maternal specialists, and other subspecialists involved in the care of pregnant women is essential for the establishment of viable research goals in obstetric and fetal pharmacology. The combined efforts of health professionals, clinical pharmacologists, molecular pharmacologists, molecular biologists, and geneticists will be needed to implement a comprehensive clinical, translational, and basic research program. Alternative study designs specific for obstetrics may need to be developed. There is also an urgent need to raise public awareness on the issue of the exclusion of pregnant women from drug studies. Finally, obstetricians and other health professionals should be encouraged to use evidence-based prescribing and support drug studies in pregnant women. The implementation of an integrated strategy to bridge the current gaps in knowledge of the safety and effectiveness of drugs used in pregnancy will yield far-reaching rewards and will result in major advances in the care of pregnant women and their offspring.
REFERENCES
Kearns, G.L., et al., Clinical Pharmacology: A Discipline Called to Action for Maternal and Child Health. Clin Pharmacol Ther, 2007. 81(4): p. 463-468. |
|
Zajicek, A. and G.P. Giacoia, Obstetric clinical pharmacology: Coming of age. Clinical Pharmacology and Therapeutics, 2007. 81(4): p. 481-482. |
|
Andrew, M.A., et al., Amoxicillin pharmacokinetics in pregnant women: modeling and simulations of dosage strategies. Clin Pharmacol Ther, 2007; 81(4): p. 547-56. |
|
Kleinman CS, Nehgme RA: Cardiac arrhythmias in the human fetus. Pediatr Cardiol. 2004 May-Jun;25(3):234-51. |
|
Mirochnick M, Best BM, Stek AM, Capparelli E, Hu C. Lopinavir exposurewith an increase dose during pregnancy J Acquir Immune Defic Syndr. 2008 Nov 4. |
|
McCullough, L.B., J.H. Coverdale, and F.A. Chervenak, A comprehensive ethical framework for responsibly designing and conducting pharmacologic research that involves pregnant women. American Journal of Obstetrics and Gynecology, 2005. 193(3, Supplement 1): p. 901-907. |
|
Mattison, D. and A. Zajicek, Gaps in knowledge in treating pregnant women. Gender Medicine, 2006. 3(3): p. 169-182. |
|
Luecke, R.H., W.D. Wosilait, and J.F. Young, Mathematical representation of organ growth in the human embryo/fetus. International Journal of Bio-Medical Computing, 1995. 39(3): p. 337-347. |
|
Chesnutt AN: Physiology of normal pregnancy. Crit Care Clin. 2004 Oct;20(4):609-15. |
|
Papantoniou N, Ismailos G, Daskalakis G et al: Pharmacokinetics of oral cefatrizine in pregnant and non-pregnant women withreference to fetal distribution. Fetal Diagn Ther. 2007;22(2):100-6. Epub 2006 Nov 27. |
|
Allegaert K, van Mieghem T, Verbesselt R et al: Cefazolin pharmacokinetics in maternal plasma and amniotic fluid during pregnancy. Am J Obstet Gynecol. 2008 Nov 10. |
|
U.S. Department of Health and Human Services, Food and Drug Administration. Guidance for the study and evaluation of gender differences in the clinical evaluation of drugs. Federal Register 58(139):39406–39416, 1993 |
|
Hunt CM, Westerkam WR, Stave GM: Effect of age and gender on the activity of human hepatic CYP3A4. Biochem Pharmacol 44: 275, 1992 |
|
Carrillo JA, Benitz J: CYP1A2 activity, gender and smoking as variables influencing the toxicity of caffeine. Br J Clin Pharmacol 41: 605, 1996 |
|
Chen M, Lee SC, Moh-Jee N, Schvirman MS et al: Pharmacokinetic analysis of bioequivalence trials: Implications for sex related issues in clinical pharmacology and biopharmaceutics. Clin Pharmacol Ther 68: 5101, 2000 |
|
Walker JS, Carmody JJ: Experimental pain in healthy human subjects: Gender differences in nociception and in response to ibuprofen. Anesth Analg 86: 12572, 1998 |
|
Gear RW, Miaskowski C, Gordon NC et al: The kappa opioid nalbuphine produces gender- and dose-dependent analgesia and antianalgesia in patients with postoperative pain. Pain 83: 339, 1999 |
|
Strocchi E, Valtancoli G, Ambrosioni E, Strocchi: The incidence of cough during treatment with angiotensin converting enzyme inhibitors. J Hypertens [Suppl] 7: S308, 1989 |
|
Makkar RR, Fromm BS, Steinman RT et al: Female gender as a risk factor for torsades de pointes associated with cardiovascular drugs. JAMA 270: 2590, 1993 |
|
Devi PK, Mehta SK, Manchanda A: Vitamin B 12 and folate status in normal puerperium. Indian J Med Res 61: 54, 1973 |
|
Hayward M, Montgomery RD, Wilson CL: The xylose test in relation to folic acid status in pregnancy. J Obstet Gynaecol Br Commonw 79: 327, 1972 |
|
Rogers MC, Willerson JT, Goldblatt A et al: Serum digoxin concentration in the human fetus, neonate and infant. N Engl J Med 287: 1010, 1972 |
|
Rauramo L, Pulkkinen M, Hartiala K: Glucuronide formation in paturients. Ann Med Exp Biol Fenn 41: 32, 1963 |
|
Ramsey RE, Strauss RG, Wilder BJ: Status epilepticus in pregnancy: Effect of phenytoin malabsorption on seizure control. Neurology 28: 85, 1978 |
|
Abramson FP. The use of stable isotopes in drug metabolism studies. Semin Perinatol 25:133, 2001 |
|
Cooper LV, Stephen GW, Aggett PJA: Elimination of pethidine and bupivacaine in the newborn. Arch Dis Child 52: 638, 1977 |
|
Morselli PL, Rovei V: Placental transfer of pethidine and norpethidine. Eur J Clin Pharmacol 18: 25, 1980 |
|
Csogor S, Csutak J, Pressler A: Modifications of albumin transport capacity in pregnant women and newborn infants. Biol Neonate 13: 211, 1968 |
|
Kuhnert BR, Kuhnert PM, Tu AL et al: Meperidine and normeperidine levels following meperidine administration during labor. I. Mother. Am J Obstet Gynecol 133: 904, 1979 |
|
Robson SC, Mutch E, Boy RJ et al: Apparent liver blood flow during pregnancy: A serial study using indicyanine green clearance. Br J Obstet Gynaecol 97: 720, 1990 |
|
Bologa M, Tang B, Klein J et al: Pregnancy-induced changes in drug metabolism in epileptic women. J Pharmacol Exp Ther 257: 735, 1991 |
|
Anderson GD. Pregnancy-induced changes in pharmacokinetics. Clin Pharmacokinet 2005;44:989-1008 |
|
Anderson GD: Pregnancy-induced changes in pharmacokinetics: a mechanistic-based approach. Clin Pharmacokinet. 2005;44(10):989-1008. |
|
Hebert MF, Easterling TR, Kirby B et al: Effects of pregnancy on CYP3A and P-glycoprotein activities as measured bydisposition of midazolam and digoxin: a University of Washington specializedcenter of research study. Clin Pharmacol Ther. 2008 Aug;84(2):248-53. Epub 2008 Feb 20. |
|
Wadelius M, Darj E, Frenne G, Rane A: Induction of CYP2D6 in pregnancy. Clin Pharmacol Ther 62: 400, 1997 |
|
Crawford JS, Rudofsky S: Some alterations in the pattern of drug metabolism associated with pregnancy, oral contraceptives and the newly born. Br J Anaesth 38: 446, 1966 |
|
Lander CM, Edwards VE, Eadie MT: Plasma anticonvulsant concentrations during pregnancy. Neurology 27: 128, 1977 |
|
Bernus I, Hooper WD, Dickinson RG, Eadie MJ: Effects of pregnancy on various pathways of human antiepileptic drug metabolism. Clin Neuropharmacol 20: 13, 1997 |
|
Garland M, Abildskov KM, Taylor S et al: Fetal morphine metabolism and clearance are constant during late gestation. Drug Metab Dispos. 2006 Apr;34(4):636-46. Epub 2006 Jan 27. |
|
Garland M, Abildskov KM, Kiu TW et al: Placental transfer and fetal elimination of morphine-3-beta-glucuronide in thepregnant baboon. Drug Metab Dispos. 2008 Sep;36(9):1859-68. Epub 2008 Jun 19. |
|
Garland M, Szeto HH, Daniel SS et al: Zidovudine kinetics in the pregnant baboon. J Acquir Immune Defic Syndr Hum Retrovirol 11: 117, 1996 |
|
Jeong H, Choi S, Song JW et al: Regulation of UDP-glucuronosyltransferase (UGT) 1A1 by progesterone and itsimpact on labetalol elimination. Xenobiotica. 2008 Jan;38(1):62-75. |
|
Kovo M, Kogman N, Ovadia O et al: Carrier-mediated transport of metformin across the human placenta determined byusing the ex vivo perfusion of the placental cotyledon model. Prenat Diagn. 2008 Jun;28(6):544-8. |
|
Kovo M, Haroutiunian S, Feldman N et al: Determination of metformin transfer across the human placenta using a duallyperfused ex vivo placental cotyledon model. Eur J Obstet Gynecol Reprod Biol. 2008 Jan;136(1):29-33. Epub 2007 Mar 12. |
|
Nandakumaran M, Gardey C, Challier JC et al: Transfer of salbutamol, ritodrine and norepinephrine in the human placenta. In Mathieu H, Pontonnier G, Olive G (eds): Pharmacologie du Developpement. Paris, Institut National de la Sante et de la Recherche Medicalis, 1980 |
|
Liggins GC, Vaugham GS: Intravenous infusion of salbutamol in the treatment of premature labor. J Obstet Gynaecol Br Commonw 80: 29, 1973 |
|
Ganapathy V, Prasad PD, Ganapathy ME, Leibach FH: Placental transporters relevant to drug distribution across the maternal—fetal interface. J Pharmacol Exp Ther 294: 413, 2000 |
|
Lankas GR, Wise LD, Cartwright ME et al: Placental P-glycoprotein deficiency enhances susceptibility to chemically induced birth defects in mice. Reprod Toxicol 12: 457, 1998 |
|
Unadkat JD, Dahlin A, Vijay S: Placental drug transporters. Curr Drug Metab. 2004 Feb;5(1):125-31. |
|
Wang RJ, Newport DJ,Stowe ZN, Donovan et al. The emrgent importance of transport proteins in the psychopharmacological treatment of the pregnant patient. Drug Metab Rev 2007:723-746 |
|
Pollex E, Lubetsky A, Koren G: The role of placental breast cancer resistance protein in the efflux of glyburideacross the human placenta. Placenta. 2008 Aug;29(8):743-7. Epub 2008 Jun 16. |
|
Weir N,He,SM, Li XT, Wang et al. Placenta Drug Metabolism and its clinical implications. Curr Drug Metab 2008;106-121 |
|
Stevens MW, Harbison RD: Placental transfer of diphenylhydantoin effects of species, gestational age and route of administration. Teratology 9: 317, 1974 |
|
Hakkola J, Pelkonen O, Pasanen M, Raunio H: Xenobiotic-metabolizing cytochrome P450 enzymes in the human feto-placental unit: Role in intrauterine toxicity. Crit Rev Toxicol 28: 35, 1998 |
|
Pelkonen O: Transplacental transfer of foreign compounds and their metabolism by the fetus. In Bridges JW, Chasseaud LL (eds): Progress in Drug Metabolism, p 119. London, John Wiley & Sons, 1977 |
|
Pelkonen O: Environmental influences on human fetal and placental xenobiotic metabolism. Eur J Clin Pharmacol 18: 17, 1980 |
|
Pelkonen O, Saarni H: Unusual patterns of benzo [a] pyrene metabolites and DNA benzo [a] pyrene adducts produced by human placental microsomes in vitro. Chem Biol Interact 30: 287, 1980 |
|
Juchav MR: Placental drug metabolism with respect to transplacental carcinogenesis. In Reznik-Schuller HM (ed): Comparative Perinatal Carcinogenesis. Boca Raton, Florida, CRC Press, 1984 |
|
Wakai RT, Strasburger JF, Li Z et al: Magnetocardiographic rhythm patterns at initiation and termination of fetalsupraventricular tachycardia. Circulation. 2003 Jan 21;107(2):307-12. |
|
Rein AJ, O'Donnell C, Geva T et al: Use of tissue velocity imaging in the diagnosis of fetal cardiac arrhythmias. Circulation. 2002 Oct 1;106(14):1827-33. |
|
Ehrnebo M, Agurell S, Jalling B, Boreus LO: Age differences in drug binding by plasma proteins: Studies on human fetuses, neonates and adults. J Clin Pharmacol 3: 189, 1971 |
|
Kurz H, Mauser-Ganshorn A, Stickel HH: Differences in the binding of drugs to plasma proteins from newborn and adult man. Eur J Clin Pharmacol 11: 463, 1977 |
|
Szeto HH, Mann LI, Bhakthavathsalan A: Meperidine pharmacokinetics in the maternal fetal unit. J Pharmacol Exp Ther 206: 448, 1978 |
|
Pelkonen O: Biotransformation of xenobiotics in the fetus. Pharmacol Ther 10: 261, 1980 |
|
Yaffe SY, Rane Am Sjoquist F et al: The presence of monooxygenase system in the human fetal liver microsomes. Life Sci 9: 1189, 1970 |
|
Treluyer JM, Gueret G, Cheron G et al: Developmental expression of CYP2C and CYP2C-dependent activities in the human liver: in vivo/in vitro correlation and inducibility. Pharmacogenetics 7: 441, 1997 |
|
Pasanen M, Pelkonen O, Kauppila A et al: Characterization of human fetal hepatic cytochrome P450. Dev Pharmacol Ther 10: 125, 1987 |
|
Komori M, Nishio K, Kitada M et al: Fetus-specific expression of a form of cytochrome P-450 in human livers. Biochemistry 29: 4430, 1990 |
|
Brazier JL, Renaud H, Ribon B, Salle BL: Plasma xanthine levels in low birth weight infants treated or not treated with theophylline. Arch Dis Child 54: 194, 1979 |
|
de Wildt SN, Kearns GL, Leeder JS, van den Anker JN: Glucuronidation in humans: Pharmacogenetic and developmental aspects. Clin Pharmacokinet 36: 439, 1999 |
|
Garland M, Szeto HH, Daniel SS et al: Saturation kinetics of morphine glucuronidation in the fetal baboon. Pediatr Res 47: 470A, 2000 |
|
Ring JA, Ghabrial H, Ching MS et al: Fetal hepatic drug elimination. Pharmacol Ther 84: 429, 1999 |
|
Garland M, Myers MM, Szeto HH, Stark RI: Maternal-fetal pharmacokinetics: The two compartment model revisited. Pediatr Res 39: 72A, 1996 |
|
Metzler M, McLachlan JA: Oxidative metabolism of diethylstilbestrol and steroidal estrogens as a potential factor in their fetotoxicity. In Neubert D, Merker HJ, Nau H, Langman J (eds): Role of Pharmacokinetics in Prenatal and Perinatal Toxicology, p 123. Stuttgart, Georg Thieme Verlag, 1977 |
|
Pelkonen O: Studies on Drug Metabolizing Enzymes in the Human Fetus. Academic Thesis, University of Oulund. |
|
Sereni F, Mandelli M, Principi N et al: Induction of drug metabolizing enzyme activities in the human fetus and in the newborn infant. Enzyme 15: 318, 1973 |
|
Rane A, Garle M, Borga O, Sjoquist F: Plasma disappearance of transplacentally transferred phenytoin in the newborn studied with mass fragmentography. Clin Pharmacol Ther 15: 39, 1974 |
|
Bray RE, Boe RW, Johnson WL: Transfer of ampicillin into fetus and amniotic fluid from maternal plasma in late pregnancy. Am J Obstet Gynecol 96: 938, 1966 |
|
Seeds AE, Kock HC, Myers RE et al: Changes in Rhesus monkey amniotic fluid pH, pCO2, and bicarbonate concentration following maternal and fetal hypercarbia and fetal death in utero. Am J Obstet Gynecol 97: 67, 1967 |
|
Duenhoelter JH, Pritchard JA: Fetal respiration: Quantitative measurements of amniotic fluid inspired near term by human and rhesus monkey. Am J Obstet Gynecol 125: 306, 1976 |
|
Landau R: Pharmacogenetics and obstetric anesthesia. Anesthesiol Clin. 2008 Mar;26(1):183-95, viii-ix. |
|
Labeling for Human Prescription Drug and Biological Products; FDA Requirements for Pregnancy and Lactation Labeling, CFR 201; Docket No 2006-N-0515-0001. Proposed Rule-making |
|
Little BB: Pharmacokinetics during pregnancy: Evidence-based maternal dose formulation. Obstet Gynecol 93: 858, 1999 |
|
Jusko WJ, Ko HC, Ebling WF: Convergence of direct and indirect pharmacodynamic response models. J Pharmacokinet Biopharm 23: 5, 1995 |
|
Wyska EB, Jusko WC: Approaches to pharmacokinetic and pharmacodynamic modeling during pregnancy. Semin Perinatol 25: 124, 2001 |
|
Smiley RM, Finster M. Do receptors get pregnant too? Adrenergic receptor alterations in human pregnancy. J Maternal Fetal Med 5:106, 1996 |
|
Guay J, Grenier Y, Varin F: Clinical pharmacokinetics of neuromuscular relaxants in pregnancy. Clin Pharmacokinet 34: 483, 1998 |
|
Hagstedt S, Rane A: Plasma concentration-effect relationship of metoprolol during and after pregnancy. Eur J Clin Pharmacol 44: 243, 1993 |
|
Knight AH, Rhind EG: Epilepsy and pregnancy: A study of 153 pregnancies in 59 patients. Epilepsia 16: 99, 1975 |
|
Weissman MM, Olfson M: Depression in women: Implications for health care research. Science 269: 799, 1995 |
|
Schatz M: Interrelationships between asthma and pregnancy: A literature review. J Allergy Clin Immunol 103: S330, 1990 |
|
Gilstrap LC 3rd, Cunningham FG, Whalley PJ: Acute pyelonephritis in pregnancy: An anterorespective study. Obstet Gynecol 57: 409, 1981 |
|
Luxford AME, Kellaway GSM: Pharmacokinetics of digoxin in pregnancy. Eur J Clin Pharmacol 25: 117, 1983 |
|
Koren G, Farine D, Maresky D et al: Significance of the endogenous digoxin-like substance found in infants and mothers. Clin Pharmacol Ther 36: 759, 1984 |
|
Lang RM, Borrow KM: Pregnancy and heart disease. Clin Perinatol 12: 551, 1985 |
|
Elkayam V: Antiarrhytmics in pregnancy and lactation. Clin Pharmacokinet 25: 254, 1987 |
|
Coppage KH, Sibai BM: Treatment of hypertensive complications in pregnancy. Curr Pharm Des. 2005;11(6):749-57. |
|
Denolle T, Weber JL, Calvez C et al: Diagnosis of white coat hypertension in pregnant women with teletransmitted homeblood pressure. Hypertens Pregnancy. 2008;27(3):305-13. |
|
Sibai BM. Treatment of hypertension in pregnant women. N Engl J Med 335:257, 1996 |
|
Ferrer RL, Sibai BM, Murlow CD et al: Management of mild chronic hypertension during pregnancy. A review. Obstet Gynecol 96: 849, 2000 |
|
Magee LA, Ornstein MP, von Dadelszen P: Management of hypertension in pregnancy. Br Med J 318: 1332, 1999 |
|
Rogers RC, Sibai BM, Whybren WD: Labetalol pharmacokinetics in pregnancy-induced hypertension. Am J Obstet Gynecol 162: 362, 1990 |
|
Rubin PC, Butters L, Kelman AW et al: Labetalol disposition and concentration effect relationships during pregnancy. J Clin Pharmacolol 15: 465, 1983 |
|
Saotome T, Minoura S, Terashi K et al: Labetalol in hypertension during the third trimester of pregnancy: Its antihypertensive effect and pharmacokinetic-dynamic analysis. J Clin Pharmacol 33: 979, 1993 |
|
Hogstedt S, Lindberg B, Peng DR et al: Pregnancy-induced increase in metoprolol metabolism. Clin Pharmacol Ther 37: 688, 1985 |
|
Middelkoop CM, Dekker GA, Kraayenbrink AA, Popp-Snijders C: Platelet-poor plasma serotonin in normal and preeclamptic pregnancy. Clin Chem 39: 1675, 1993 |
|
Bolte AC, van Geijn HP, Dekker GA: Pharmacological treatment of severe hypertension in pregnancy and the role of serotonin-receptor blockers. Eur J Obstetr Gynecol Reprod Biol 95: 22, 2001 |
|
The Seventh Report of the Joint National Committee on Prevention,Detection, Evaluation, and Treatment of High Blood Pressure. National Heart, Lung and Blood Institute. August 2004 |
|
Boutroy MJ: Fetal and neonatal effects of the beta adrenoceptor blocking agents. Dev Pharmacol Ther 10: 224, 1987 |
|
Mabie WC, Gonzalez AR, Sibai B: A comparative trial of labetalol and hydralazine in the acute management of severe hypertension complicating pregnancy. Obstet Gynecol 70: 328, 1987 |
|
Plouin PF, Breart G, Maillard F et al: Comparison of antihypertensive efficacy and perinatal safety of labetalol andmethyldopa in the treatment of hypertension in pregnancy: a randomized controlledtrial. Br J Obstet Gynaecol. 1988 Sep;95(9):868-76. |
|
el-Qarmalawi AM, Morsy AH, al-Fadly A et al: Labetalol vs. methyldopa in the treatment of pregnancy-induced hypertension. Int J Gynaecol Obstet. 1995 May;49(2):125-30. |
|
Caton AR, Bell EM, Druschel CM at al. Maternal hypertension, antihypertensive use and the risk of severe hypospadias. Birth Defects Res A Clin Mol Teratol 2008;82; 34-40 |
|
Montella KR. Pulmonary pharmacology in pregnancy. Clin Chest Med 13:587, 1992 |
|
Rayburn WF, Atkinson BD, Gilbert K, Turnbull GL: Short-term effects of inhaled albuterol on maternal and fetal circulations. Am J Obstet Gynecol 171: 770, 1994 |
|
Juniper EF, Newhouse MT: Effect of pregnancy on asthma: A critical appraisal of the literature. In Schatz M, Zeiger RS: Asthma and Allergy in Pregnancy and Early Infancy, pp 223–249. New York, Marcel Dekker, 1993 |
|
Bakhireva LN, Schatz M, Jones KL et al: Asthma control during pregnancy and the risk of preterm delivery or impairedfetal growth. Ann Allergy Asthma Immunol. 2008 Aug;101(2):137-43. |
|
Barron WM, Left AR. Asthma in pregnancy. Am Rev Respir Dis 147:510, 1993 |
|
Dombrowski MP, Schatz M, Wise R et al: Randomized trial of inhaled beclomethasone dipropionate versus theophylline formoderate asthma during pregnancy. Am J Obstet Gynecol. 2004 Mar;190(3):737-44. |
|
Osur SL: The management of asthma and rhinitis during pregnancy. J Womens Health (Larchmt). 2005 Apr;14(3):263-76. |
|
Park-Wyllie L, Mazzotta P, Pastuszak A et al: Birth defects after maternal exposure to corticosteroids: Prospective cohort study and meta-analysis of epidemiological studies. Teratology 62: 385, 2000 |
|
Metzger BE, Buchanan TA, Coustan DR et al: Summary and recommendations of the Fifth International Workshop-Conference onGestational Diabetes Mellitus. Diabetes Care. 2007 Jul;30 Suppl 2:S251-60. |
|
Klieger C, Pollex E, Koren G: Treating the mother--protecting the unborn: the safety of hypoglycemic drugs inpregnancy. J Matern Fetal Neonatal Med. 2008 Mar;21(3):191-6. |
|
Zhou L, Naraharisetti SB, Wang H et al: The breast cancer resistance protein (Bcrp1/Abcg2) limits fetal distribution ofNetwork and University of Washington Specialized Center of Research Study. Mol Pharmacol. 2008 Mar;73(3):949-59. Epub 2007 Dec 13. |
|
Nicholson W, Bolen S, Witkop CT et al: Benefits and Risks of Oral Diabetes Agents Compared With Insulin in Women WithGestational Diabetes: A Systematic Review. Obstet Gynecol. 2009 Jan;113(1):193-205. |
|
Kimber-Trojnar Z, Marciniak B, Leszczynska-Gorzelak B et al: Glyburide for the treatment of gestational diabetes mellitus. Pharmacol Rep. 2008 May-Jun;60(3):308-18. |
|
Goldberg HL, Nossim R: Psychotropic drugs in pregnancy and lactation. Intl J Psychiatry Med 24: 129, 1994 |
|
Nars PW, Girard J: Lithium carbonate intake during pregnancy leading to large goiter in a premature infant. Am J Dis Child 131: 924, 1977 |
|
Morell P, Sutherland GR, Baumah PK et al: Lithium toxicity in a newborn. Arch Dis Child 58: 539, 1983 |
|
Kozma C: Neonatal toxicity and transient neurodevelopmental deficits following prenatalexposure to lithium: Another clinical report and a review of the literature. Am J Med Genet A. 2005 Feb 1;132(4):441-4. |
|
Rey E, Giraux P, d'Athis P et al: Pharmacokinetics of the placental transfer and distribution of clorazepate and its metabolite nordiazepam in the feto-placental unit and in the neonate. Eur J Clin Pharmacol 15: 181, 1979 |
|
Wisner KL, Perel JM, Wheeler SB: Tricyclic dose requirements across pregnancy. Am J Psychiatry 150: 1541, 1993 |
|
Belik J: Fetal and neonatal effects of maternal drug treatment for depression. Semin Perinatol. 2008 Oct;32(5):350-4. |
|
Hostetter A, Ritchie JC, Stowe ZN: Amniotic fluid and umbilical cord blood concentrations of antidepressants in three women. Biol Psychiatry 48: 1032, 2000 |
|
Rumack BH, Walvarens PA: Neonatal withdrawal following maternal ingestion of ethclorvynol (Placidyl). Pediatrics 52: 714, 1973 |
|
Goetz RL, Bain RV: Neonatal withdrawal symptoms associated with maternal use of pentazocine. J Pediatr 84: 887, 1974 |
|
Athinarayanan P, Peirog SH, Nigam SK, Glass L: Chlordiazepoxide withdrawal in the neonate. Am J Obstet Gynecol 124: 212, 1976 |
|
Altshuler LL, Cohen L, Szuba MP et al: Pharmacologic management of psychiatric illness during pregnancy: Dilemmas and guidelines. Am J Psychiatry 153: 592, 1996 |
|
Meador KJ, Pennell PB, Harden CL et al: Pregnancy registries in epilepsy: a consensus statement on health outcomes. Neurology. 2008 Sep 30;71(14):1109-17. Epub 2008 Aug 13. |
|
Perucca E, Crema A: Plasma protein binding of drugs in pregnancy. Clin Pharmacokinet 7: 336, 1982 |
|
Yerby MS, Friel PN, McCormick K et al: Pharmacokinetics of anticonvulsants in pregnancy: alterations in plasma protein binding. Epilepsy Res 5: 223, 1990 |
|
Hanson JW, Smith DW: The fetal hydantoin syndrome. J Pediatr 87: 285, 1975 |
|
Feldman GL, Weaver DD, Lourien EW: The fetal trimethadione syndrome: Report of an additional family and further delineation of this syndrome. Am J Dis Child 131: 1389, 1977 |
|
Yerby MS The use of anticonvulsants during pregnancy. Semin Perinatol 25:153, 2001 |
|
Allen RW, Ogden B, Bentley FL, Jung AL: Fetal hydantoin syndrome, neuroblastoma and hemorrhagic disease in a neonate. JAMA 244: 1464, 1980 |
|
Atkinson DE, Brice-Bennett S, D'Souza SW: Antiepileptic medication during pregnancy: does fetal genotype affect outcome? Pediatr Res. 2007 Aug;62(2):120-7. |
|
Dansky L, Andermann E, Rosenblatt D et al: Anticonvulsant folate levels and pregnancy outcome: A prospective study. Ann Neurol 21: 176, 1987 |
|
Mosley BS, Cleves MA, Siega-Riz AM et al: Neural tube defects and maternal folate intake among pregnancies conceived afterfolic acid fortification in the United States. Am J Epidemiol. 2009 Jan 1;169(1):9-17. Epub 2008 Oct 25. |
|
Davis VA, Rothberg AD, Argent AC et al: Precursor thrombin status in patients receiving anticonvulsant drugs. Lancet 1: 126, 1985 |
|
Cerveny L, Pavek P, Malakova J et al: Lack of interactions between breast cancer resistance protein (bcrp/abcg2) andselected antiepileptic agents. Epilepsia. 2006 Mar;47(3):461-8. |
|
Knott C, Williams CP, Reynolds F: Phenytoin kinetics during pregnancy and the puerperium. Br J Obstet Gynaecol 93: 1030, 1986 |
|
Green DM, Fine WE, Lit P: Offspring of patients treated for unilateral Wilms' tumor in childhood. Cancer 49: 2285, 1982 |
|
Selevan SG, Lindbohm ML, Hornung RW et al: A study of occupational exposure to antineoplastic drugs and fetal loss in nurses. N Engl J Med 313: 1173, 1985 |
|
Gokal R, Durrant J, Bennet JD: Successful pregnancy in acute monocytic leukemia. Br J Cancer 34: 299, 1976 |
|
Finau SA: Pregnancy in Di Guglielmo's syndrome. Med J Aust 1: 320, 1980 |
|
Hamer JW, Beard ME, Doff GB: Pregnancy complicated by acute myeloid leukemia. NZ Med J 89: 212, 1979 |
|
Lowenthal RM, Marsden KA, Newman NM et al: Normal infant after treatment of acute myeloid leukemia in pregnancy with daunorubicin. Aust NZ J Med 8: 431, 1978 |
|
Lilleyman JS, Hill AS, Anderson KJ: Consequences of acute myelogenous leukemia in early pregnancy. Cancer 40: 1300, 1977 |
|
Durie BGM, Giles HR: Successful treatment of acute leukemia during pregnancy. Arch Intern Med 90: 137, 1977 |
|
Lenhard MS, Bauerfeind I, Untch M: Breast cancer and pregnancy: challenges of chemotherapy. Crit Rev Oncol Hematol. 2008 Sep;67(3):196-203. Epub 2008 Apr 3. |
|
Davison JM, Dellagrammatikas H, Parkin JM: Maternal azathioprine therapy and depressed haemopoiesis in the babies of renal allograft patients. Br J Obstet Gynaecol 92: 233, 1985 |
|
Keskin N, Yilmaz S: Pregnancy and tuberculosis: to assess tuberculosis cases in pregnancy in adeveloping region retrospectively and two case reports. Arch Gynecol Obstet. 2008 Nov;278(5):451-5. Epub 2008 Feb 14. |
|
Holdiness MR: Teratology of the antituberculosis drugs: A review. Early Hum Dev 15: 61, 1987 |
|
Holdiness MR: Neurological manifestations and toxicities of the antituberculosis drugs: A review. Med Toxicol 2: 33, 1987 |
|
Sterling Y, Woolf L, North WRD et al: Hemostasis in normal pregnancy. Thromb Haemost 52: 176, 1984 |
|
Chunilal SD, Young E, Johnston M et al: Comparing the in vivo response to heparin in the plasma of non-pregnant and pregnant women. Blood 92 (Suppl): 359A, 1998 |
|
Zimran A, Shilo S, Fisher D: Histomorphometric evaluation of reversible heparin-induced osteoporosis in pregnancy. Arch Intern Med 146: 386, 1986 |
|
Douketis JD, Ginsberg JS, Burrows RF et al: The effects of long-term heparin therapy during pregnancy on bone density. A prospective matched cohort study. Thromb Haemost 75: 254, 1996 |
|
Hirsch J, Warkentin TE, Raschke et al: Heparin and low-molecular heparin: mechanisms of action, pharmacokinetics and safety. Chest 114: 489S, 1998 |
|
Casele HL, Laifer SA, Woelkers DA, Venkataramanan R: Changes in the pharmacokinetics of the low-molecular-weight heparin enoxaparin sodium during pregnancy. Am J Obstet Gynecol 181: 1113, 1999 |
|
Deruelle P, Coulon C: The use of low-molecular-weight heparins in pregnancy--how safe are they? Curr Opin Obstet Gynecol. 2007 Dec;19(6):573-7. |
|
Chan WS, Anand S, Ginsberg JS: Anticoagulation of pregnant women with mechanical heart valves: a systematic review of the literature. Arch Intern Med 160: 191, 2000 |
|
Hall JG, Pauli RM, Wilson K: Maternal and fetal sequelae of anticoagulation during pregnancy. Am J Med 68: 122, 1980 |
|
Chang MKW, Harvey D, DeSwiet M: Follow-up study of children whose mothers were treated with warfarin during pregnancy. Br J Obstet Gynaecol 91: 70, 1984 |
|
Marchant B, Brownlie BEW, Hart DM et al: The placental transfer of propylthiouracil, methimazole, and carbimazole. J Clin Endocrinol Metab 45: 1187, 1977 |
|
Chan GW, Mandel SJ: Therapy insight: management of Graves' disease during pregnancy. Nat Clin Pract Endocrinol Metab. 2007 Jun;3(6):470-8. |
|
O'Allemand A, Gruters A, Heidemann F, Schurnbrand P: Iodine-induced alterations of thyroid function in newborn infants after prenatal and perinatal exposure to povidone—iodine. J Pediatr 102: 935, 1983 |
|
Milham S, Elledge W: Maternal methimazole and congenital defects in children. Teratology 5: 125, 1972 |
|
Refetoff S, Ochi Y, Selenkow HA, Rosenfeld RL: Neonatal hypothyroidism and goiter in one infant of each of two sets of twins due to maternal therapy with antithyroid drugs. J Pediatr 85: 240, 1974 |
|
Kyriazopoulou V, Michalaki M, Georgopoulos N et al: Recommendations for thyroxin therapy during pregnancy. Expert Opin Pharmacother. 2008 Feb;9(3):421-7. |
|
Korzeniowski OM: Antibacterial agents in pregnancy. Infect Dis Clin North Am 9: 639, 1995 |
|
Philipson A: Plasma levels of ampicillin in pregnant women following administration of ampicillin and pivampicillin. Am J Obstet Gynecol 130: 674, 1978 |
|
Philipson A: Pharmacokinetics of antibiotics in pregnancy and labour. Clin Pharmacokinet 4: 297, 1979 |
|
Ripamonti D, Cattaneo D, Maggiolo F et al: Atazanavir plus low-dose ritonavir in pregnancy: pharmacokinetics and placentaltransfer. AIDS. 2007 Nov 30;21(18):2409-15. |
|
Pacifici GM: Placental transfer of antibiotics administered to the mother: a review. Int J Clin Pharmacol Ther. 2006 Feb;44(2):57-63. |
|
Burr CK, Lampe MA, Corle S et al: An end to perinatal HIV: success in the US requires ongoing and innovativeefforts that should expand globally. J Public Health Policy. 2007 Jul;28(2):249-60. |
|
Stek AM, Mirochnick M, Capparelli E et al: Reduced lopinavir exposure during pregnancy. AIDS. 2006 Oct 3;20(15):1931-9. |
|
O'Sullivan MJ, Boyer PJ, Scott GB et al: The pharmacokinetics and safety of zidovudine in the third trimester o pregnancy for women infected with human immunodeficiency virus and their infants: phase I acquired immunodeficiency syndrome clinical trials group study (protocol 082). Zidovudine Collaborative Working Group. Am J Obstet Gynecol 168: 1510, 1993 |
|
O'Sullivan MJ, Boyer PJ, Scott GB et al: The pharmacokinetics and safety of zidovudine in the third trimester o pregnancy for women infected with human immunodeficiency virus and their infants: phase I acquired immunodeficiency syndrome clinical trials group study (protocol 082). Zidovudine Collaborative Working Group. Am J Obstet Gynecol 168: 1510, 1993 |
|
Blanche S, Tardieu M, Rustin P et al: Persistent mitochondrial dysfunction and perinatal exposure to antiretroviral nucleoside analogues. Lancet 354: 1084, 1999 |
|
Morselli PL, Rovei V: Placenta transfer of pethidine and nor-pethidine and their pharmacokinetics in the newborn. Eur J Clin Pharmacol 18: 25, 1980 |
|
Albright GA, Joyce TH III, Stevenson DK: Systemic medication. In Albright GA, Ferguson JE II, Joyce TH III, Stevenson DK (eds): Anesthesia in Obstetrics: Maternal, Fetal and Neonatal Aspects, p 165. Boston, Butterworths, 1986 |
|
Morselli PL, Morselli RF, Bossi L: Clinical pharmacokinetics in newborn and infants. Clin Pharmacokinet 5: 485, 1980 |
|
Stambaugh JE, Wainer IW, Schwartz S: The effect of phenobarbital on the metabolism of meperidine in normal volunteers. J Clin Pharmacol 18: 482, 1978 |
|
Committee on Drugs: Naloxone use in newborns. Pediatrics 65:667, 1980 |
|
Goodlin RC: Naloxone and its possible relationship to fetal endorphin levels and fetal distress. Am J Obstet Gynecol 139: 16, 1981 |
|
Hatjis LG, Meis PJ: Sinusoidal fetal heart rate pattern associated with butorphanol administration. Obstet Gynecol 67: 377, 1986 |
|
Evron S, Ezri T: Options for systemic labor analgesia. Curr Opin Anaesthesiol. 2007 Jun;20(3):181-5. |
|
Tucker GT, Mather LE: Pharmacokinetics of local anesthetic agents. Br J Anaesth 47: 213, 1975 |
|
Magno A, Berlin A, Karrlson K et al: Anesthesia for cesarean section: Intravenous placental transfer and elimination of bupivacaine following epidural analgesia for elective cesarean section. Acta Anaesthesiol Scand 20: 141, 1976 |
|
Kuhnert B, Kuhnert P, Reese A et al: Maternal and neonatal eliminators of CABA after epidural anesthesia with 2-chloroprocaine during paturition. Anesth Analg 61: 638, 1982 |
|
Tucker GT, Mather LE: Clinical pharmacokinetics of local anesthetics. Clin Pharmacokinet 4: 241, 1979 |
|
Biehl D, Shnider SM, Levinson G et al: Placental transfer of lidocaine: Effects of fetal acidosis. Anesthesiology 48: 409, 1978 |
|
Ralston DH, Schnider SM: The fetal and neonatal effects of regional anesthesia in obstetrics. Anesthesiology 48: 34, 1978 |
|
Kuhnert BR, Harrison MJ, Linn PL, Kuhnert PM: Effects of maternal epidural anesthesia on neonatal behavior. Anesth Analg 63: 301, 1984 |
|
Baden JM, Rice SA, Serra M et al: Thynidine and methiomine synthesis in pregnant rats exposed to nitrous oxide. Anesth Analg 62: 738, 1983 |
|
Tsatsaris V, Cabrol D, Carbonne B: Pharmacokinetics of tocolytic agents. Clin Pharmacokinet. 2004;43(13):833-44. |
|
Papatsonis DN, Bos JM, van Geijn HP et al: Nifedipine pharmacokinetics and plasma levels in the management of preterm labor. Am J Ther. 2007 Jul-Aug;14(4):346-50. |
|
Gross TL, Kuhnert BR, Kuhnert PM et al: Maternal and fetal plasma concentrations of ritodrine. Obstet Gynecol 65: 793, 1985 |
|
Caritas SN, Toig G, Heddinger LA, Ashmead GA: A double blind study comparing ritodrine and terbutaline in the treatment of preterm labor. Am J Obstet Gynecol (Suppl 1) 150: 7, 1984 |
|
Caldwell G, Scougall I, Boddy K, Toft AD: Fasting hyperinsulinemic hypoglycemia after ritodrine therapy for premature labor. Obstet Gynecol 70: 478, 1987 |
|
Kazzi NJ, Gross TL, Kazzi GM, Williams TG: Neonatal complications following in utero exposure to intravenous ritodrine. Acta Obstet Gynecol Scand 66: 65, 1987 |
|
Hansen NB, Oh W, Larochelle F, Stonestreet BS: Effects of maternal ritodrine administration in neonatal renal function. J Pediatr 103: 774, 1983 |
|
Herrera AJ, Macaraeg AL: Maternal ritodrine hydrochloride therapy associated with transient leukemoid reaction in the infant. South Med J 79: 78, 1986 |
|
Doyle LW, Crowther CA, Middleton P et al: Magnesium sulphate for women at risk of preterm birth for neuroprotection of thefetus. Cochrane Database Syst Rev. 2007 Jul 18;(3):CD004661. |
|
Zuckerman H, Shalev E, Gilad G, Katzuni E: Further study of the inhibition of premature labor by indomethacin. J Perinat Med 12: 19, 1984 |
|
Read W, Wellby DE: The use of a calcium antagonist (nifedipine) to suppress preterm labor. Br J Obstet Gynaecol 93: 933, 1986 |
|
Simhan HN, Caritis SN: Prevention of preterm delivery. N Engl J Med. 2007 Aug 2;357(5):477-87. |
|
Meis PJ, Klebanoff M, Thom E et al: Prevention of recurrent preterm delivery by 17 alpha-hydroxyprogesteronecaproate. N Engl J Med. 2003 Jun 12;348(24):2379-85. |
|
Attardi BJ, Zeleznik A, Simhan H et al: Comparison of progesterone and glucocorticoid receptor binding and stimulation ofrelated progestins. Am J Obstet Gynecol. 2007 Dec;197(6):599.e1-7. |
|
Boylan P: Oxytocic drugs in labor. In Lewis P (ed): Clinical Pharmacology in Obstetrics, pp 324–336. Boston, Wright PSG, 1983 |
|
Seitchik J, Amico J, Robinson AB, Castillo M: Oxytocin augmentation of dysfunctional labor: IV oxytocin pharmacokinetics. Am J Obstet Gynecol 150: 225, 1984 |
|
Lange AP, Secher NJ, Westergaard JG, Skovgard IL: Neonatal jaundice after labour induced or stimulated by prostaglandin E2 or oxytocin. Lancet 1: 991, 1982 |
|
Turner JE, Shannon B, Porreco RP, Weiss MA: Prostaglandin E2 in tylose gel for cervical ripening before induction of labor. J Reprod Med 32: 815, 1987 |
|
Facchinetti F, Venturini P, Fazzio M et al: Elective cervical ripening in women beyond the 290th day of pregnancy: arandomized trial comparing 2 dinoprostone preparations. J Reprod Med. 2007 Oct;52(10):945-9. |
|
Ballard PL, Ballard RA: Corticosteroids and respiratory syndrome status. Pediatrics 63: 163, 1979 |
|
Antenatal Corticosteroids Revisited: Repeat Courses. NIH Consensus Statement. On line August 17–18, 2000 |
|
Murphy KE, Hannah ME, Willan AR et al: Multiple courses of antenatal corticosteroids for preterm birth (MACS): arandomised controlled trial. Lancet. 2009 Dec 20;372(9656):2143-51. |
|
Coustan DR: Antenatal enhancement of pulmonary maturation. Clin Perinatol 14: 697, 1987 |
|
Kwong MS, Egan EA: Reduced incidence of hyaline membrane disease in extremely premature infants following delay of delivery in mother with preterm labor. Pediatrics 78: 767, 1986 |
|
Shaywitz SE, Cohen DJ, Shaywitz BA: Behavior and learning difficulties in children of normal intelligence born to alcoholic mothers. J Pediatr 96: 978, 1980 |
|
Rossett HL, Ovellette EM, Weiner L, Owens E: Therapy of heavy drinking during pregnancy. Obstet Gynecol 51: 41, 1978 |
|
Wright JT, Barrison IG, Lewis IG et al: Alcohol consumption, pregnancy and low birthweight. Lancet 1: 663, 1983 |
|
Halmesmaki E, Raivio KO, Ylikorkala O: Patterns of alcohol consumption during pregnancy. Obstet Gynecol 69: 594, 1987 |
|
Krose J, LeFevre M. Zweig S: Changes in smoking and alcohol consumption during pregnancy: A population-based study in a rural area. Obstet Gynecol 67: 627, 1986 |
|
Bearer CF, Stoler JM, Cook JD et al: Biomarkers of alcohol use in pregnancy. Alcohol Res Health. 2004-2005;28(1):38-43. |
|
Longo LD: The biological effects of carbon monoxide on the pregnant woman, fetus and newborn infant. Am J Obstet Gynecol 129: 69, 1977 |
|
Mebert A, Sande H, Foss OP, Stenwig JT: Smoking during pregnancy: Effects on the fetus and on thiocyanate levels in mother and baby. Acta Paediatr Scand 68: 547, 1979 |
|
Luck W, Nau H, Hansen R, Steldinger R: Extent of nicotine and cotinine transfer to the human fetus, placenta, and amniotic fluid of smoking mothers. Dev Pharmacol Ther 8: 384, 1985 |
|
Kelly J, Matthews KA, O'Conor M: Smoking in pregnancy: Effects on mother and fetus. Br J Obstet Gynaecol 91: 111, 1984 |
|
Pace HB, Davis WM, Borgen LA: Teratogenesis and marijuana. Ann NY Acad Sci 191: 123, 1971 |
|
Greenland S, Staisch KJ, Brown N, Gross SJ: The effects of marijuana use during pregnancy: A preliminary epidemiologic study. Am J Obstet Gynecol 143: 408, 1982 |
|
Linn S, Schoenbaum SC, Monson RR et al: The association of marijuana use with outcome in pregnancy. Am J Public Health 73: 1161, 1983 |
|
Quzi QH, Mariano E, Milman DH et al: Abnormalities in offspring associated with prenatal marijuana exposure. Dev Pharmacol Ther 8: 141, 1985 |
|
Chasnoff IJ, Hatcher R, Burns WJ, Schnoll SH: Pentazocine and triplenamine (T's and blues) effects on the fetus and neonate. Dev Pharmacol Ther 6: 162, 1983 |
|
Cox S, Posner SF, Kourtis AP, Jamieson DJ. Hospitalizations with amphetamine abuse among pregnant women. Obstet Gynecol 2008;111:341-7 |
|
Oro As, Dixon SD: Perinatal cocaine and methamphetamine exposure: Maternal and neonatal correlates. J Pediatr 111: 571, 1987 |
|
Chasnoff IJ, Bussey M, Savich R, Stack CM: Perinatal infarction and maternal cocaine use. J Pediatr 108: 456, 1986 |
|
Bingol N, Fuchs M, Diaz V et al: Teratogenicity of cocaine in humans. J Pediatr 110: 93, 1987 |
|
Chasnoff IJ, Burns WJ, Hatcher RP, Burns KA: Phencyclidine: Effects on the fetus and neonate. Dev Pharmacol Ther 6: 404, 1983 |
|
Ranzini A, Allen A, Lai Y: Use of complementary medicines and therapies among obstetric patients. Obstet Gynecol 97: S46, 2001 |
|
Friedman JM: Teratology Society: Presentation to the FDA public meeting on safety issues associated with the use of dietary supplements during pregnancy. Teratology 62: 134, 2000 |
|
Jones TK, Lawson BM: Profound neonatal congestive heart failure caused by maternal consumption of blue cohosh herbal medication. J Pediatr 132: 550, 1998 |
|
Wright IM: Neonatal effects of maternal consumption of blue cohosh. J Pediatr 134: 384, 1999 |
|
Chan E. Displacement of bilirubin from albumin by berberine. Biol Neonate 63:201, 1993 |
|
Cvan Engelen AD, Weijtens O, Brenner JI et al: Management outcome and follow-up of fetal tachycardia. J Am Coll Cardiol 24: 1371, 1994 |
|
Ito S. Transplacental treatment of fetal tachycardia: Implications of drug transporting proteins in the placental. Semin Perinatol 25: 196, 2001 |
|
Forrest M, David M: Prenatal treatment of congenital adrenal hyperplasia due to 21-hydrolase deficiency. Seventh International Congress of Endocrinology, No, 911, Quebec Canada, 1984 |
|
Rayburn WF, Farmer KC: Off-label prescribing during pregnancy. Obstet Gynecol Clin North Am. 1997 Sep;24(3):471-8. |
|
Chauvenet M, Rimailho A, Hoog-Labouret N: Methodology for the evaluation of drugs in pregnant women. Therapie. 2003 May-Jun;58(3):247-58. |
|
Anger GJ, Piquette-Miller M: Translational pharmacology: harnessing increased specialization of researchwithin the basic biological sciences. Clin Pharmacol Ther. 2008 Jun;83(6):797-801. |
|
Coverdale JH, McCullough LB, Chervenak FA: The ethics of randomized placebo-controlled trials of antidepressants withpregnant women: a systematic review. Obstet Gynecol. 2008 Dec;112(6):1361-8. |
|
NIH Guide for Grants. RFA HD-03-17. July 29, 2003. Reissued as RFA HD 09-002 December 16, 2008 |
|
Sharma S, Ou J, Strom S et al: Identification of enzymes involved in the metabolism ofpreterm birth. Drug Metab Dispos. 2008 Sep;36(9):1896-902. Epub 2008 Jun 23. |
|
Ravindran S, Zharikova OL, Hill RA et al: Identification of glyburide metabolites formed by hepatic and placentalmicrosomes of humans and baboons. Biochem Pharmacol. 2006 Dec 15;72(12):1730-7. Epub 2006 Aug 30. |
|
Nanovskaya TN, Patrikeeva S, Hemauer S et al: Effect of albumin on transplacental transfer and distribution of rosiglitazoneand glyburide. J Matern Fetal Neonatal Med. 2008 Mar;21(3):197-207. |
|
Dickmann LJ, Tay S, Senn TD et al: Changes in maternal liver Cyp2c and Cyp2d expression and activity during ratpregnancy. Biochem Pharmacol. 2008 Apr 15;75(8):1677-87. Epub 2008 Feb 7. |
|
Zharikova OL, Ravindran S, Nanovskaya TN et al: Kinetics of glyburide metabolism by hepatic and placental microsomes of human and baboon. Biochem Pharmacol. 2007 Jun 15;73(12):2012-9. Epub 2007 Mar 12. |
|
Hernandez-Diaz S, Hernan MA, Meyer K et al: Case-crossover and case-time-control designs in birth defects epidemiology. Am J Epidemiol. 2003 Aug 15;158(4):385-91. |
|
Toh S, Mitchell AA, Louik C et al: Selective Serotonin Reuptake Inhibitor Use and Risk of Gestational Hypertension.selective serotonin reuptake inhibitors (SSRIs) on the risks of gestational hypertension.Am J Psychiatry. 2009 Jan 2. |
|
Louik C, Lin AE, Werler MM et al: First-trimester use of selective serotonin-reuptake inhibitors and the risk ofbirth defects. N Engl J Med. 2007 Jun 28;356(26):2675-83. |
|
Slikker W Jr, Young JF, Corley RA et al: Improving predictive modeling in pediatric drug development: pharmacokinetics, pharmacodynamics, and mechanistic modeling. Ann N Y Acad Sci. 2005 Aug;1053:505-18. |
|
Horgan RP, Clancy OH, Myers JE et al: An overview of proteomic and metabolomic technologies and their application to pregnancy research. BJOG. 2009 Jan;116(2):173-81. |