Ovarian Dysgenesis and Premature Ovarian Failure Caused by X Chromosomal Abnormalities
Authors
INTRODUCTION
Ovarian failure exists in a variety of forms and has a variety of causes. Failure may be complete or premature, occurring earlier (younger than 40 years) than the expected age of menopause. Multiple etiologies exist. Mendelian and polygenic forms are discussed by Simpson in the chapter "XX Gonadal Dysgenesis and Premature Ovarian Failure (POF) in 46,XX Individuals", and teratogenic forms by Verp in the chapter "Environmental Factors Causing Ovarian Failure in Humans". In this chapter discussion is restricted to ovarian failure caused by absence of perturbations of the X chromosome. This chapter inevitably reflects the author’s previous publications on the topic.1, 2, 3, 4, 5, 6, 7
REPRODUCTIVE EMBRYOLOGY
Primordial germ cells originate in the endoderm of the yolk sac and migrate to the genital ridge to form the indifferent gonad, which is initially indistinguishable in 46,XY and 46,XX embryos. Primordial germ cells migrate from the yolk sac around 28 days (postconception), reaching the gonadal ridge by 37 days, or 5 days after formation of the genital ridge. Indifferent gonads develop into testes if the embryo – or more specifically the gonadal stroma – is 46,XY. This process begins about 43 days after conception (15 mm crown–rump length (CRL)). Testes become morphologically identifiable 7–8 weeks after conception (9–10 weeks' gestational or menstrual weeks).
Sertoli cells are the first cells to become recognizable in testicular differentiation, then organizing the surrounding cells into tubules. Both Leydig cells8 and Sertoli cells9 differentiate prior to testicular morphogenesis, consistent with their directing gonadal development rather than the converse. These two cells secrete hormones that direct different aspects of male differentiation (Fig. 1).
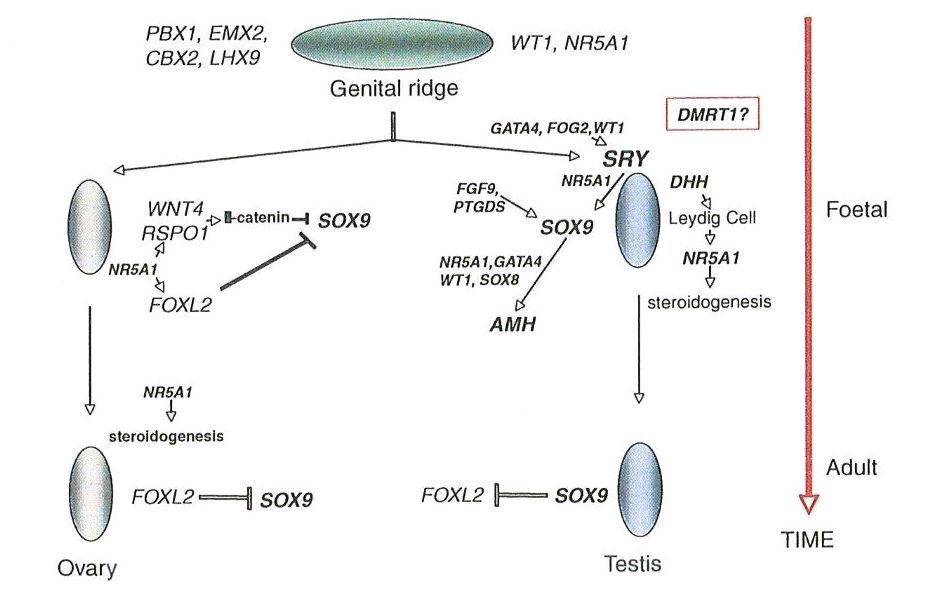 Fig. 1. Schematic diagram illustrating embryonic genes involved in gonadal differentiation in the normal female. (From Simpson JL: Disorders of the gonads, genital tract and genitalia. In: Rimoin DL, Connor JM, Pyeritz RE, Korf BR, eds. Principles and Practice of Medical Genetics, 6th edn., New York, Churchill Livingston, 2012. In press.)
Fig. 1. Schematic diagram illustrating embryonic genes involved in gonadal differentiation in the normal female. (From Simpson JL: Disorders of the gonads, genital tract and genitalia. In: Rimoin DL, Connor JM, Pyeritz RE, Korf BR, eds. Principles and Practice of Medical Genetics, 6th edn., New York, Churchill Livingston, 2012. In press.)
Fetal Leydig cells, first recognized at 55 days, produce by 63 days an androgen (testosterone) that stabilizes Wolffian ducts and permits differentiation of the vasa deferentia, epididymides, and seminal vesicles. Following conversion of testosterone by 5α-reductase to dihydrotestosterone (DHT), external genetalia are virilized. These actions can be mimicked by the administration of testosterone to female or castrated male mammalian embryos. Fetal Sertoli cells produce antimüllerian hormone (AMH, previously called müllerian inhibitory substance or MIS), a glycoprotein that diffuses locally to cause regression of müllerian derivatives (uterus and fallopian tubes). AMH also has functions related to gonadal development, given that if AMH is chronically expressed in XX transgenic mice, oocytes fail to persist, tubule-like structures develop into gonads, and müllerian differentiation is abnormal.10 Wolffian differentiation (vasa differentiation, epididymides, seminal vesicles) require testosterone.
In the absence of a Y chromosome, the indifferent gonad develops into an ovary11, 12 at least embryologically. Transformation into fetal ovaries begins at 50–55 days of embryonic development. By 20 weeks of embryonic life fetal ovaries contain up to 7 million germ cells, the vast majority undergoing atresia with only 400 actually ovulated as adults. In 46,X embryos, oocytes differentiate only to undergo atresia at a rate more rapid than that occurring in normal 46,XX embryos. Thus, genes on the X necessary for normal development actually control ovarian maintenance. These genes are localized to specific regions of the X, and must be accompanied by various autosomal genes to generate normal ovaries.
Ductal and external genital development occur independent of gonadal differentiation because in the absence of testosterone and AMH, external and internal genitalia develop in female fashion. In the absence of AMH, müllerian ducts form the uterus and fallopian tubes, and wolffian ducts regress. Such changes occur in normal XX embryos, as well as in XY animals that were castrated as embryos prior to testicular differentiation.
Importantly, germ cells are present in 45,X embryos.13 This is well demonstrated by their presence of germ cells in 45,X abortuses, which account for 10% of all first trimester abortions (see Simpson and Carson, Chapter "Genetic and nongenetic causes of pregnancy loss"). Pathogenesis of germ cell absence in 45,X adults thus involves atresia occurring at a rate more rapid than that occurring in normal 46,XX embryos. Pathogenesis does not involve failure of germ cell formation. This explains the occasional pregnancy in 45,X women (see below). In fact, germ cells are present in all monosomy X mammals (e.g., mice), and often in adults as well. If two intact X chromosomes are required to prevent 45,X ovarian follicles from degenerating prematurely, the second X chromosome must be responsible for ovarian maintenance, rather than primary ovarian differentiation.
The specific mechanism appears to involve expression of WNT4 and RSPO1, both autosomal loci. If SRY is lacking, SOX9 remains repressed14 preventing male differentiation. Continued repression of SOX9 is accomplished by FOXL2.15 FOXL2 is thus essential for ovarian development. Many other genes – X chromosome and autosomal – are, however, necessary for normal ovarian development to proceed normally.
OVARIAN FAILURE AS RESULT OF MONOSOMY X (TURNER SYNDROME)
Historically, the term applied to women with ovarian failure is gonadal dysgenesis or Turner syndrome. Turner syndrome is a broad term, however, and here the term gonadal dysgenesis is applied to women with streak gonads and the term Turner stigmata is used for those having short stature and certain somatic anomalies (Table 1). By themselves Turner stigmata as defined would not imply the presence of streak gonads. The term Turner syndrome would be applied to those individual with both streak gonads, Turner stigmata, and a 45,X or X-deletion complement.
Table 1. Somatic features associated with the 45,X chromosomal complement
Growth
Decreased birth weight
Decreased adult height (141–146 cm)
Intellectual function
Verbal IQ higher than performance IQ
Cognitive deficits (space-form blindness)
Immature personality, probably secondary to short stature
Craniofacial
Premature fusion sphenoccipital and other sutures, producing brachycephaly
Abnormal pinnae
Retruded mandible
Ptosis
Hypertelorism
Epicanthal folds (25%)
High-arched palate (36%)
Abnormal dentition
Visual anomalies, usually strabismus (22%), anbtyopia
Hypernetropia auditory deficits; sensorineural or secondary to middle ear infection
Neck
Pterygium colli (46%)
Short, broad neck (74%)
Low nuchal hair (71%)
Chest
Rectangular contour (shield chest) (35%)
Apparent widely spaced nipples
Tapered lateral ends of clavicle
Cardiovascular
Coarctation of aorta or ventricular septal defect (10–16%)
Renal (38%)
Horseshoe kidneys
Unilateral renal aplasia
Duplication ureters
Gastrointestinal
Telangiectasias
Skin and lymphatics
Pigmented nevi (63%)
Lymphedema (38%), generalized, caused by hypoplasia superficial vessels; puffy hands and
feet
Nails
Hypoplasia and malformation (66%)
Skeletal
Cubitus valgus (54%)
Radial tilt of articular surface of trochlear
Clinodactyly V
Short metacarpals, usually IV (48%)
Decreased carpal arch (mean angle 117degree)
Deformities of medical tibial condyle
Dermatoglyphics
Increased total digital ridge count
Increased distance between palmar triradii a and b
Distal axial triradius in position t
Percentages affected reflect tabulation of Simpson, from which table is modified.16
Cytologic origin
In 80% of 45,X cases the X is maternal (Xm) in origin. In the remaining 20%, the remaining X is paternal (Xp) in origin.17, 18
Gonads
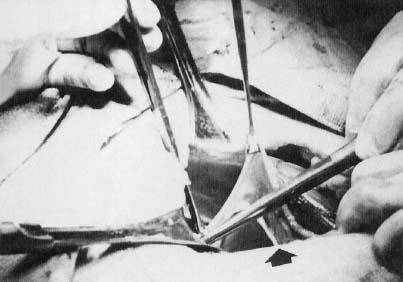
In monosomy X the gonad usually exists not as the typical ovoid structure but as a white fibrosis streak, 2–3 cm long and approximately 0.5 cm wide, located in the position ordinarily occupied by the ovary (Fig. 2). A streak gonad is characterized histologically by interfacing waves of dense fibrosis stroma (Fig. 3).
|
|
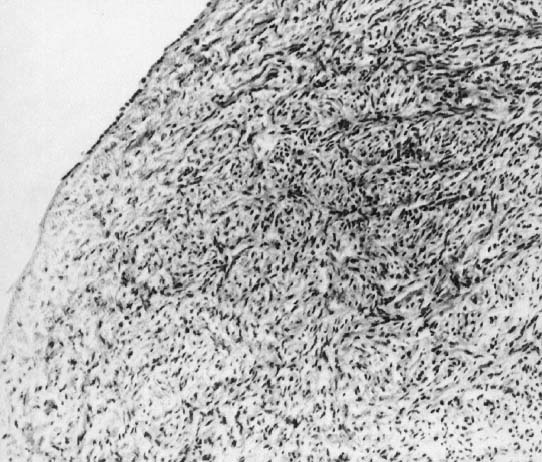
That 45,X individuals show streak gonads as adults is not as obvious as might be predicted, given that relatively normal ovarian development occurs in many other mammals (e.g., mice) with monosomy X. The presumptive explanation is that pivotal genes on the normal heterochromatic (inactive) X are being inactivated. Of the some 2000 genes on the X, only perhaps 5% escape inactivation.19 Most of these genes are on the X short arm (Xp), clustered in selected euchromatic regions. Candidate genes for ovarian maintenance genes will probably prove to lie in these euchromatic regions.
The endocrinologic correlates of ovarian failure are deficient secretion of sex steroids. Estrogen and androgen levels are thus decreased; follicle-stimulating hormone (FSH) and luteinizing hormone (LH) levels are compensatively increased. Deficiencies of estrogen-dependent processes leads to predictable effects of hormonal deficiency. Premenarchal uterine enlargement and growth spurt are not observed. Pubic and axillary hair fail to develop at puberty (Fig. 4). Breasts contain little parenchymal tissue, and areolar tissue is only slightly darker than the surrounding skin. External genitalia, vagina, and müllerian derivatives are well differentiated, but remain small (unstimulated) in the absence of exogenous steroids.
|
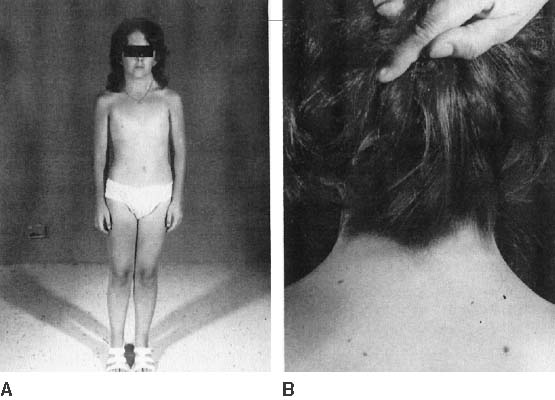
Approximately 3–5% of adult 45,X patients menstruate spontaneously (at least twice) and show breast development. Many fertile patients have been reported, as recently reviewed by Abir and colleagues13 and Hovatta.20 An undetected 46,XX cell line (i.e., 45,X/46,XX mosaicism) should be suspected in menstruating 45,X patients. This is especially plausible in reports like that of Magee and colleagues21 who observed seven pregnancies in one ostensibly 45,X woman. However, it is not expected that some 45,X individuals could be fertile, in as much as germ cells are present in 45,X embryos. In addition, pregnancy can occasionally be achieved in hypergonadotropic women by sequential gonadotropin suppression followed by ovulation induction. Check and colleagues22 induced ovulation in 5% of 361 cycles in 100 hypergonadotropic women; although their chromosomal complements were not stated.
Hormonal treatment of 45,X women usually involves hormone therapy (estrogen and cyclic progestogens). This can result in normal uterine size, which could be followed by assisted reproductive technology (ART) if pregnancy is desired. The process involves a partner’s sperm being mixed with ova donated from another woman, fertilization in vitro, and transfer of embryo to the uterus of the hormonally synchronized 45,X patient. The success rate (clinical pregnancies) is over 20% per cycle. For example, Foudila and colleagues23 reported 20 clinical pregnancies among 18 women with Turner’s syndrome, the exact chromosomal complements not being stated. Although the clinical pregnancy rate per fresh embryo transferred was impressive at 46% (13/28), seven of the 13 (54%) resulted in spontaneous abortion, for a take-home baby rate of 46% (6/13). Among transferred frozen embryos the rates were 28% (7/25) and 14% (1/7), respectively. Similar pregnancy rates were also reported by Khastgir and colleagues24 and Tarani and colleagues.25 The ability of a 45,X woman to carry her pregnancy to term must be addressed before embarking on ART. Specifically, women with coarctation of the aorta may be unsuitable candidates. The increased prevalence of autoimmune disease (e.g., thyroiditis), carbohydrate dysfunction (diabetes mellitus), hypertension, and other adult-onset diseases further places many 45,X patients in high-risk situations.
Offspring of 45,X women show little, if any, increased risk for chromosomal abnormalities.26, 27 Claims to the contrary13 need to be tempered by biases of biases of ascertainment not being taken into account. Outcomes are relatively normal in pregnancies observed after the index case was diagnosed (truncate analysis). Adverse outcomes prior to that time were probably the reason cytogenetic studies were initiated. Spontaneous abortions are increased in donor oocyte ART, but this probably reflects hormonal dysfunction or uterine factors (hypoplasia) rather than transmission of aneuploid (monosomic) gametes.
Somatic anomalies
45,X individuals not only are short (less than 4 feet 10 inches) but often exhibit various somatic anomalies (Turner stigmata) (Table 1). No single feature(s) of Turner stigmata is pathognomonic, but in aggregate a characteristic spectrum exists that is more likely to exist in 45,X individuals than in individuals having most other sex chromosomal abnormalities. Systematic evaluation of renal, vertebral, cardiac, and auditory function is obligatory, irrespective of the patient’s age when diagnosed.
Growth
45,X neonates are usually low birth weight. Total body length at birth is less than normal, but often close to the 50th centile. Before puberty, height velocity falls in the 10th–15th centile,28 and the mean height of untreated 45,X adults (younger than 16 years old) is between 141 and 146 cm,16, 29 perhaps 20 cm less than normal. In normal females the predicted adult height can be estimated by summing the heights of both parents, dividing by 2, and subtracting by 13 cm.30 Taking into account decreased expected height for 45,X individuals, correlation of the height of a 45,X offspring with midparental height holds for Turner syndrome as it does for normal 46,XX females.31 That is, absolute height predicted in Turner syndrome is less but the midparental height correlation still holds.
Various treatments for short stature in 45,X patients have been proposed, including growth hormone (GH), anabolic steroids, and low-dose estrogen.32 Most treatment regimens show ostensible benefit, especially immediately after onset of therapy. Consensus is now that the ultimate height can be increased by 6–8 cm by GH treatment alone.33, 34 Current treatment is human recombinant DNA-derived human GH. GH results in an 8.4 ± 4.5 cm increase in height over that predicted; final height was 150.4 ± 5.5 cm in one heterogeneous group.35 With GH and oxandrolone, the increase was 10.3 ± 4.7 cm. Treatment regimens are discussed in standard pediatric endocrinologic treatises (e.g., Grumbach and Conte36). In general, treatment is begun at 2–5 years of age, and stopped at approximately 15 years of age.37 Low-dose estrogen is deferred until final height is near;35 high-dose estrogen should then be given to stimulate secondary sexual development. The latter regimen should begin at 14–15 years of age, starting with 0.3–0.625 mg conjugated equine estrogens daily for 6–12 months; dose is then increased to 1.25 mg.
A potential reason for limited efficacy of growth hormone treatment may be that epiphyses in 45,X individuals are structurally abnormal. Not only long bones, but teeth38 and skull39 are also abnormal. Thus, patients with a 45,X chromosomal complement could be said to have a skeletal dysplasia.
Some wonder whether the presence of an unappreciated Y-bearing line (e.g., 45,X/46,XY) could lead to neoplastic consequences with treatment. However, few ostensibly nonmosaic 45,X cases will prove to have a 46,XY even after analysis of thousands of cells using fluorescent in situ hybridization (FISH) with a Y probe.
Intelligence
Most 45,X patients are of normal intelligence, but any given patient has a higher probability of being retarded than a 46,XX person.16 Performance IQ is lower than verbal IQ, the latter being similar to 46,XX matched controls. 45,X individuals may have a cognitive defect characterized by poor spatial processing skills (space-form blindness). Ross and colleagues40 opined that only loss of distal Xp (Xp 22.33) produced this phenotype; regions responsible for neurocognitive deficits were believed to be distant from statural or ovarian abnormalities.
Psychosocial deficits primarily reflect behavioral immaturity and difficulties in social relationships. These are probably secondary to delayed sexual development and statural growth.41, 42 The possibility has been raised that parental origin influences phenotype Xm (X of maternal origin). Xm cases are said to show cognitive deficits more often than Xp.43 If true, this would indicate the X contains imprinted genes.
Adult-onset diseases
Many adult-onset disorders occur in 45,X cases with frequencies greater than expected in the general population. Hypertension deserves special comment, given its presence in about one third of adult 45,X individuals. Hypertension need not alter hormonal therapy; however, careful monitoring is required, and exogenous estrogen therapy may need to be reduced. Frequencies of diabetes mellitus and autoimmune thyroiditis are increased.
Is a particular region on the X responsible for somatic anomalies in 45,X?
The region of the X responsible for somatic anomalies in 45,X, that if deleted results in somatic anomalies, is unclear, but it is not necessarily the same as that for ovarian maintenance. The distal X short arm (Xp) has in particular been implicated in somatic development. A pseudoautosomal gene implicated in somatic development and short stature (Turner stigmata) is SHOX. This pseudoautosomal locus is not subject to inactivation. RPS4X is another candidate for the same reason. Zinn and colleagues44 and Zinn and Ross45 have attempted to correlate somatic anomalies with Xp perturbations, using molecular markers to define deletions and X/autosomal translocation. High-arched palate, short stature, and autoimmune thyroid disease were associated with terminal deletions of Xp11.2-22.1, the same region noted to contain ovarian determinants. Boucher and colleagues46 concluded that Xp11.4 is critical for lymphoedema. Bioné and Toniolo47 have also discussed candidate genes on Xp and Xq that could be important for somatic differentiation.
45,X/46,XX AND 45,X/47,XXX MOSAICISM
If nondisjunction or anaphase lag occurs in the zygote or embryo, two or more cell lines may result (mosaicism). The final chromosomal complement depends on the stage at which abnormal cell division occurs and the types of daughter cells that survive after the abnormal cell division (nondisjunction). Ability to detect mosaicism depends on the number of cells analyzed per tissue and the number of tissues analyzed. For example, counting 50 cells without detecting one nonmodal cell excludes (p < 0.005) mosaicism for a minority line of 10% or more.48 The common practice of a cytogenetic laboratory counting 20 cells carries a probability of 0.122 for failing to detect at least one cell representing a minority cell line of 10% frequency. With 50 cells, probability falls to <0.005. If the minority cell line constituted 30%, the probability of failing to detect would be <0.001 if 20 cells were counted.48
The most common form of mosaicism associated with ovarian failure or gonadal dysgenesis is 45,X/46,XX. Population-based data do not exist, but it seems that 45,X/46,XX individuals show fewer anomalies than 45,X individuals. One survey revealed 12% of 45,X/46,XX individuals menstruate, compared with only 3% of 45,X individuals.16 Among 45,X/46,XX individuals in that survey, 18% underwent breast development, compared with 5% of 45,X individuals. Mean adult height is greater in 45,X/46,XX than in 45,X; more mosaic (25%) than nonmosaic (5%) patients reach adult heights greater than 162 cm.16 Somatic anomalies are less likely to exist in 45,X/46,XX than in 45,X. 45,X/47,XXX is less common than but phenotypically similar to 45,X/46,XX individuals.
Some 45,X/46,XY individuals may also show bilateral streak gonads; however, they often show a unilateral streak gonad and a contralateral dysgenetic testis (mixed gonadal dysgenesis) or bilateral testes. This phenotype is discussed in the chapter "Cytogenetic, teratogenic, and miscellaneous other disorders causing male pseudohermaphroditism or germ cell failure in both males (46,XY) and females (46,XX)".
STRUCTURAL ABNORMALITIES OF THE X CHROMOSOME
X-Deletions
Cytogenetically obvious (>6 Mb) structural abnormalities of the X have long been recognized. Different terminal deletions of the short arm of the X chromosome exist, reflecting different amounts of persisting Xp. Breakpoints presumably do not occur in just selected places, but rather throughout the chromosome. However, pooling structurally similar terminal deletions reveals the most common breakpoint for terminal deletions to be Xp11.2 → 11.4. In 46,X,del(X)(p11) only proximal Xp remains; the del(Xp) chromosome thus appears acrocentric or telocentric. More distal (telomeric) breakpoints also exist: Xp21, 22.1, 22.2, 22.3. Sequencing and analysis with polymorphic DNA markers are beginning to permit precise determinations of these breakpoints, but still relatively little molecular information exists.44, 45, 49, 50
In 45,X,del(Xp)(p11) most of Xp is missing. Approximately half of these individuals show primary amenorrhea and gonadal dysgenesis. In a tabulation made 15 years ago, 12 of 27 reported del(X)(p11.2 → 11.4) individuals menstruated spontaneously; however, menstruation was rarely normal.4 More recent calculations1, 2, 51 have not materially altered the fundamental conclusions concerning location of key determinants. Figure 5 shows a compilation of cases reported through 1999. Similar conclusions were reached by Ogata and Matsuo,52 who estimate that 50% of del(Xp11) cases show primary amenorrhea and 45% show secondary amenorrhea. Both Wandstrat and colleagues53 and Thomas and Huson54 were impressed by single nonmosaic cases del(X) (p11.2) cases showing normal height and fertility.
|
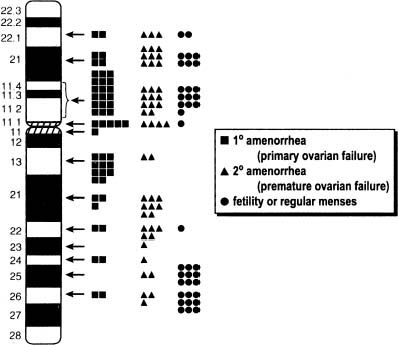
Women with more distal Xр deletions [del(X)(p21.1 to p22.122)] menstruate more often, but many are still infertile or show secondary amenorrhea. Ovarian failure (menopause) is more likely to be premature (younger than 35 or 40 years of age, depending on definition). Thus, Xp21, 22.1 or 22.2 is less important for ovarian development1, 51 than Xp11. Deletion of only the most telomeric portion of Xp (Xp22.3 → Xpter) does not result in amenorrhea.55 Zinn and colleagues44 and Zinn and Ross45 concluded that only the region Xp11.3 → 22.1 was paramount, although this large region encompasses most of Xp. Cases with an interstitial deletion will be necessary to narrow the region of interest.44, 45 Interstitial deletions have involved Xp11-22 and Xp11.4-22.3.56, 57
Familial cases have been reported. These include both mother and daughter showing the same Xp-deletions49, 58 as well as X/autosome translocations. Among ten del(Xp) cases studied by James and colleagues59 were two mother-daughter pairs. A later analysis reported 25 females with de novo deletions of Xp and four familial Xp deletions.54 Familial cases have involved deletions at Xp11 as well as Xp22-12.44, 60
X-ISOCHROMOSOME
If division of the centromere occurs in the transverse rather than the longitudinal plane, an isochromosome results. The resulting metacentric chromosome consists of isologous arms. Both arms are structurally identical and contain the same genes. Isochromosome for the X long arm [i(Xq)] differs from terminal deletion of Xp in that not just the terminal portion but all of the Xp is deleted. An isochromosome for the X long arm is the most common X structural abnormality, but coexisting 45,X cells (mosaicism) are common.
Almost all reported 46,i(Xq) patients show streak gonads, and short stature, and Turner’s stigmata have long been accepted as almost universal.16 Only rarely do 46,X,i(Xq) patients seem to menstruate. The more often complete lack of gonadal development in 46,X,i(Xq) individuals contrasts with that in 46,X,del(Xp11) individuals, about half of whom menstruate or develop breasts (see above). This could indicate that gonadal determinants exist at several different locations on Xp. That is, a locus near the centromere could be deleted in i(Xq) yet retained in del(X)(p11).
OVARIAN GENES ON Xp AND Xq
Although gene designations have as noted been assigned to the chromosomal regions on Xp and Xq (POF1, POF2), the actual genes remain unclear. Array CGH copy number variants (CNV), which are microdeletions smaller than detectable by conventional karyotype, are found in women with ovarian failure but not consistently correlated with the regions expected if genes were restricted to regions assigned for POF1 and POF2. Knauff et al.61 found CNVs (262 kb resolution deletions) in Xq21.3 to be associated with premature ovarian failure but in a region where PCHCHIIX and TGHF2LX are located. However, no deletions were found in other regions considered on the basis of cytogenetic studies to be pivotal. Quilter et al.62 found that 48% of their cases showed a CNV microduplication or deletion with many X regions involved, whereas Dudding et al.63 found only a 800 kb duplication at Xq13.3 and a Xp22.3 duplication among 50 POF cases (4%). Aboura et al.64 reported a statistically significant association for a 0.217 kb gain on Xq28, but the same gain was found in 5/67 controls. No significant X-deletions were found in 90 POF cases. Discrepancies among these CNV studies are most likely explained by failure to perform the parental array studies that are necessary to exclude paternal transmission of a polymorphic CNV of no likely clinical significance.
Genome-wide linkage analysis has shown association between Xp21.3 (LOD score 3.1) and the age of menopause.65 Ochalski66 recently proposed a list of genes having potential relevance to ovarian development and function, some of which receive specific comment below.
CANDIDATE GENES ON Xp
Deletions and other structural abnormalities of the X chromosome (short arm or long arm) are associated with ovarian failure, either complete or partial. The phenotype is discussed elsewhere67, 68 and genes on Xp must be pivotal for ovarian germ cell function. Here, as elsewhere6, 7we review selected candidate genes.
USP9X (ubiqulitin-specific protease 9)
This gene maps to Xp11.4, a pivotal region.69 In Drosophila USP9X is required for eye development and oogenesis. The role USP9X plays in human gonadal development is still unclear, but its location in the important Xp11.4 region is tantalizing.
ZFX (zinc finger X)
ZFX, a DNA binding protein, maps to Xp22.1-21.3. It is homologous to ZFY, once a candidate gene for male sex determination. Mice null for ZFX are small, less viable, less fertile, and characterized by diminished germ cell number in ovaries and testes.70 Their external and internal genetalia are otherwise normal.
Bone morphogenetic protein (BMP15)
The strongest candidate gene on Xp is bone morphogenetic protein 15 (BMP), a member of the transforming growth factor-beta (TGFβ) superfamily. TGF genes direct many developmental pathways through binding and activating transmembrane serine/theonine kinase receptors. BMP is involved in folliculogenesis and embryonic development, being expressed in gonads. BMP15 is located in Xp11.2 and consists of only two exons. Before informative human cases were reported, animal studies had suggested that perturbations of BMP15 could be important in ovarian failure. Heterozygous Inverdale sheep carrying a mutation in the BMP15 gene show an increased ovulation rate, with twin and triplet births; primary ovarian failure occurs in ovine homozygotes. Bmp15 knockout mutant female mice are subfertile, showing decreased ovulation rates, reduced litter size and decreased number of litters per lifetime .71 In humans BMP was implicated in POF first by Di Pasquale et al. who reported two sisters with POF who were heterozygous for c.704A>G substitution.72 The proband had streak gonads and elevated follicle-stimulating hormone (FSH) (80 MIU/ml); the younger sib had one episode of vaginal spotting but otherwise similar. The mother was homozygously normal at this allele, Y235C transmitted from their father. There was in vitro evidence for a dominant negative mechanism.
Among 79 American and 87 European caucasian women with POF73 nine had one of four different non-synonymous sequence variants; only one was found in control women. Among 203 European caucasian women, Laissue et al.74 reported three non-synonymous variants in 13 women with POF; only one variant was present in the control sample.74 Dixit et al.75 sequenced 202 Indian women with ovarian failure and found 18 (9%) had heterozygous BMP15 variants (SNPs). These perturbations involved 11 different non-synonymous sequences; none were present in controls. However, many novel SNPs in the POF cases were often found in only a single POF subject, raising doubts about causation.
If heterozygous BMP15 mutations exert a deleterious effect, the mutation must exert a dominant negative effect. The basis of a dominant negative effect might involve disturbed dimerization of TGFβ proteins. BMP15 protein not only forms homodimers, but also heterodimers with the GDF9 protein, an autosomal TGFβ protein.
Short stature homeobox (SHOX)
This gene lies in the pesudoautosomal region (PAR1), a regional obligatory recombination that involves Xp22 and Yp11.3 (pseudoautosomal regions X and Y that contain homologous sequences and hence, undergo synapsis during male meiosis.) In this region SHOX, a gene involved in skeletal growth. This was initially reasoned on the basis of short stature existing in Turner syndrome (45,X) but tall stature in polysomy (47,XXY). As would be expected, SHOX escapes X-inactivation, because the gene is in the pseudoautosomal region. SHOX mutation and deletions have been associated with short stature,76, 77 Leri-Weill syndrome,78 and Langer mesomelic dysplasia;79 duplications have also been observed. Tachdjian et al.80 studied three POF cases known by conventional karyotype to have an Xq deletion; a del(X)q21.31 case also had an array CGH 620 kb duplication (Xp22.3→PAR1).
Although postulated, a relationship between POF and SHOX seems unlikely. Any gene in the pseudoautosomal region is unlikely to play a major role in ovarian (or testicular) determinants because recombination would occur frequently (and obligatorily). The locus would, perhaps frequently, be lost to female offspring.
CANDIDATE GENES ON Xq
XIST (X inactivation specific transcript)
Xq13 contains the X-inactivation center and XIST. Loss of germ cells may or may not be the direct result of perturbation of XIST, despite disturbances of X-inactivation per se clearly leading to ovarian failure. The concept of a defined "critical region" necessary for ovarian development receives less attention than in the past, but is doubtless rich in pivotal genes.
DIAPH2 (diaphanous)
This candidate gene lies in the Xq21-24 region. Diaphanous (DIAPH2) is the homolog of Drosophila melanogaster diaphanous (dia). This gene family helps to establish cell polarity, govern cytokinesis, and reorganize the actin cytoskeleton. In both male and female Drosphila, dia causes sterility. Disruption of the last intron of DIAPH281 was observed in Xq21/autosome translocation associated with ovarian failure.
DACH2
DACH1, which escapes X-inactivation, was identified through an X; autosomal translocation identified through a patient with POF.82, 83 DACH2 has a Drosphila homologue and is expressed in multiple tissues, but not overtly in the reproductive system. Although five heterozygous DACH missense mutations were found in a series of 257 Italian POF cases, controls (N = 110) sometimes showed the same mutation. Thus, causality remains lacking. That DACH2 and POF1B (see below) are only 700 kb apart and in a gene-poor region raises the possibility of existence of a regulatory region that could be perturbed.
POF1B
The belief that "critical regions" for ovarian function existed on Xq originally led to the proposal that there existed POF1 and POF2, the former an ovarian region (gene?) on Xq21-ter and the later on Xq13.3-21.1. Later an X autosomal translocation led to another proposed candidate gene, called POIFB and localized to Xq2183. Lacombe84 then reported a consanguineous Lebanese family in which 46,XX sisters with POF were homozygous for a missense mutation (R329Q). Heterozygotes were unaffected, consistent with R329Q being present in the normal population. Bione et al.85 failed to find even heterozygous perturbations in over 200 POF cases.
XPNPEP2
X-propyl aminopeptidase 2, which maps to Xq25, has been proposed as relevant to the "gene" called POF2 that maps in this region. Bione and Toniolo86 and Prueitt and colleagues87 reported that XPNPEP2 was disrupted in an Xq/autosome translation associated with primary amenorrhea.
Progesterone receptor membrane component 1 (PGRMC1)
This X-linked gene was interrogated in 67 POF cases, with one heterozygous mutation found (H165R).88 The change occurred in a domain necessary for nontranscriptional regulation of cytochrome P450, potentially of functional significance.
Angiotensor II (type 2) (ATZ) receptor
This gene is expressed in fetal tissue and atretic granulosa cells in rodents. A plausible relationship can be made between this gene and POF. Its location on Xq22-23, is a region of known significance (so called POF2). Katsuya89 studied two families in each of which sibs each had POF; no AT2 mutations were found.
FMR1
Fragile X syndrome is the most common single gene cause of mental retardation in males. The disorder is the result of an increase in the number of CGG triplet repeats in a 5′ region of the X-lined gene FMR1, which is located on Xq27.3. FMR1 is a translational suppressor of a number of other genes. Hypermethylation occurs, FMR1 protein is suppressed, and other genes are secondarily overexpressed. Fragile X syndrome occurs when there are greater than 200 CGG repeats, the normal number being 29 or 30.
Approximately 15–20% of women with FMR1 premutation (55–100 CGG repeats) develop premature ovarian failure.90 Schwartz and colleagues91 found oligomenorrhea in 38% of premutation carriers versus in 6% controls. Allingham-Hawkins and coworkers92 analyzed 1268 controls, 50 familial POF cases and 244 sporadic POF cases. In this international collaborative survey92 63 of 395 premutation carriers (16%) underwent menopause before 40 years of age; the frequency in controls was 0.4%. In Atlanta women, Sullivan and colleagues93 found 12.9% of premutation carriers (N = 250;>59 repeats) had POF versus 1.3% (2/157) of controls. The number of CGG repeats significantly correlated with risk of POF but not linearly. Only a slightly increased risk exists with 40–79 repeats, whereas the risk is much higher with 80–99 repeats; no further increased risk after >100 repeats. The plateau is consistent with women having the full mutation (>200 CGG) not showing POF.92
FMR1 testing should probably be part of the work-up for premature ovarian failure, and it is so recommended in Europe.94 If oocyte or ovarian slice cryopreservation becomes more feasible, population screening might even be justified for fertility preservation.
Although located in the same region of Xq, FMRq cannot logically correspond exactly to the region which when deleted causes only ovarian failure in del(Xq). However, terminal deletions at Xq27 (karyotype or array CGH) have been observed in females with fragile X syndrome. This may be the result of skewed X-inactivation such that the sole active X is heterozygous for a FMR1 premutation transmitted from a phenotypically normal but heterozygous mother.95
REFERENCES
Simpson JL: Genetic programming in ovarian development and oogenesis. In Lobo RA, Kelsey JMR (eds): Menopause Biology and Pathobiology. pp 77, 94. London, Academic Press, 2000 |
|
Simpson JL, Rajkovic A: Ovarian differentiation and gonadal failure. Am J Med Genet 89:186, 1999 |
|
Simpson JL, Elias S: Genetics in Obstetrics and Gynecology. 3rd ed.. Philadelphia, W.B. Saunders, 2002, p. 270. |
|
Simpson, J.L.: Disorders of abnormal sexual differentiation. (In) Clinical Pediatric and Adolescent Gynecology (J.S. Sanfilippo, E. Lara-Torre, K. Edmonds, C. Templeman, eds). Informa Healthcare, New York, NY, 2009. |
|
Simpson, JL: Mammalian Sex Determination, (In) Encyclopedia of Life Sciences,John Wiley & Sons, Ltd. published online, March 2008. doi: 10.1005/9780470015902. a0001886.pub2 |
|
Simpson, JL. Disorders of the gonads, genital tract and genitalia (In) Principles and Practice of Medical Genetics, 6th ed. (Rimoin DL, Connor Jm, Pyertiz RE, Korf BR eds.), New York, Churchill-Livingstone, 2012, in press |
|
Simpson JL. Genetics of Female Fertility: X Chromosome Disorders. (In) Methods of Molecular Biology- Human Fertility (Wasserman P, Rosenwaks Z, eds.). Humana Press, New York. In press. |
|
Patsavoudi E, Magre S, Castanier M et al: Dissociation between testicular morphogenesis and functional differentiation ofLeydig cells. J Endocrinol. 1985 May;105(2):235-8. |
|
Magre S, Jost A: Dissociation between testicular organogenesis and endocrine cytodifferentiationof Sertoli cells. Proc Natl Acad Sci U S A. 1984 Dec;81(24):7831-4. |
|
Behringer RR, Cate RL, Froelick GJ et al: Abnormal sexual development in transgenic mice chronically expressing mullerianinhibiting substance. Nature. 1990 May 10;345(6271):167-70. |
|
Jost A (1953) Problems of fetal endocrinology. The gonadal and hypophyseal hormones. Recent Prog Horm Res 8:379-418. |
|
Jost A: Problems of fetal endocrinology: the adrenal glands. Recent Prog Horm Res. 1966;22:541-74. |
|
Abir R, Fisch B, Nitke S, et al: Morphological study of fully and partially isolated early human follicles. Fertil Steril 75:141, 2001 |
|
Schlessinger D, Garcia-Ortiz JE, Forabosco A et al: Determination and stability of gonadal sex. J Androl. 2010 Jan-Feb;31(1):16-25. Epub 2009 Oct 29. |
|
Uhlenhaut NH, Jakob S, Anlag K et al: Somatic sex reprogramming of adult ovaries to testes by FOXL2 ablation. Cell. 2009 Dec 11;139(6):1130-42. |
|
Simpson JL: Gonadal dysgenesis and abnormalities of the human sex chromosomes: Current status of phenotypic-karyotypic correlations. Birth Defects Orig Artic Ser 11(4):23, 1975 |
|
Mathur A, Stekol L, Schatz D, et al: The parental origin of the single X chromosome in Turner syndrome: Lack of correlation with parental age or clinical phenotype. Am J Hum Genet 48:682, 1991 |
|
Cockwell A, MacKenzie M, Youings S, et al: A cytogenetic and molecular study of a series of 45,X fetuses and their parents. J Med Genet 28:152, 1991 |
|
Willard HF: The sex chromosomes and X chromosome inactivation. In Scriver CR, Beaudet AL, Sly WS, et al (eds): The Metabolic and Molecular Bases of Inherited Disease. pp 1191, 1211 Vol 8:New York, McGraw-Hill, 2001 |
|
Hovatta O: Pregnancies in women with Turner’s syndrome. Ann Med 31:106, 1999 |
|
Magee AC, Nevin NC, Armstrong MJ, et al: Ullrich-Turner syndrome: Seven pregnancies in an apparent 45,X woman. Am J Med Genet 75:1, 1998 |
|
Check JH, Nowroozi K, Chase JS, et al: Ovulation induction and pregnancies in 100 consecutive women with hypergonadotropic amenorrhea. Fertil Steril 53:811, 1990 |
|
Foudila T, Soderstrom-Anttila V, Hovatta O: Turner’s syndrome and pregnancies after oocyte donation. Hum Reprod 14:532, 1999 |
|
Khastgir G, Abdalla H, Thomas A, et al: Oocyte donation in Turner’s syndrome: An analysis of the factors affecting the outcome. Hum Reprod 12:279, 1997 |
|
Tarani L, Lampariello S, Raguso G, et al: Pregnancy in patients with Turner’s syndrome: Six new cases and review of literature. Gynecol Endocrinol 12:83, 1998 |
|
Dewhurst J: Fertility in 47,XXX and 45,X patients. J Med Genet 15:132, 1978 |
|
Simpson JL: Pregnancies in women with chromosomal abnormalities. In In Schulman JD, Simpson JL (eds): Genetic Diseases in Pregnancy. pp 439, 471 New York, Academic Press, 1981 |
|
Brook CG, Murset G, Zachmann M, et al: Growth in children with 45,XO Turner’s syndrome. Arch Dis Child 49:789, 1974 |
|
Ranke MB, Pfluger H, Rosendahl W, et al: Turner syndrome: Spontaneous growth in 150 cases and review of the literature. Eur J Pediatr 141:81, 1983 |
|
Tanner JM, Goldstein H, Whitehouse RH: Standards for children’s height at ages 2–9 years allowing for heights of parents. Arch Dis Child 45:755, 1970 |
|
Brook CG, Gasser T, Werder EA, et al: Height correlations between parents and mature offspring in normal subjects and in subjects with Turner’s and Klinefelter’s and other syndromes. Ann Hum Biol 4:17, 1977 |
|
Ranke MB, Blum WF, Haug F, et al: Growth hormone, somatomedin levels and growth regulation in Turner’s syndrome. Acta Endocrinol 116:305, 1987 |
|
Rosenfeld RG, Grumbach MM: Turner Syndrome. New York, Marcel Dekker, 1990 |
|
Rosenfeld RG, Frane J, Attie KM, et al: Six-year results of a randomized, prospective trial of human growth hormone and oxandrolone in Turner syndrome. J Pediatr 121:49, 1992 |
|
Rosenfeld RG, Attie KM, Frane J, et al: Growth hormone therapy of Turner’s syndrome: beneficial effect on adult height. J Pediatr 132:319, 1998 |
|
Grumbach MM, Conte FA: Disorders of sex differentiation. In Wilson JD, Foster DW, Kronenberg H (eds): Williams Textbook of Endocrinology. pp. 1303, Vol. 9:Philadelphia, WB Saunders, 1998 |
|
Saenger P: Turner’s syndrome. N Engl J Med 335:1749, 1996 |
|
Filippson R, Lindsten J, Almqvist S: Time of eruption of the permanent teeth, cephalometric and tooth measurement and sulphationfactor activity in 45 patients with Turner’s syndrome with different types of X chromosome aberration. Acta Endocrinol 48:91, 1965 |
|
Lindsten J, Fraccaro M: Turner’s syndrome. In Rashad MN, Mortion WRM (eds): Genital Anomalies,. pp 396, Springfield, Charles C. Thomas Publishers, 1969 |
|
Ross JL, Roeltgen D, Kushner H, et al: The Turner syndrome-associated neurocognitive phenotype maps todistal Xp. Am J Hum Genet 67:672, 2000 |
|
McCauley E, Sybert VP, Ehrhardt AA: Psychosocial adjustment of adult women with Turner syndrome. Clin Genet 29:284, 1986 |
|
McCauley E, Kay T, Ito J, et al: The Turner syndrome: Cognitive deficits, affective discrimination, and behavior problems. Child Dev 58:464, 1987 |
|
Skuse DH, James RS, Bishop DV, et al: Evidence from Turner’s syndrome of an imprinted X-linked locus affecting cognitive function. Nature 387:705, 1997 |
|
Zinn AR, Tonk VS, Chen Z, et al: Evidence for a Turner syndrome locus or loci at Xp11.2-p22.1 Am J Hum Genet 63:1757, 1998 |
|
Zinn AR, Ross JL: Molecular analysis of genes on Xp controlling Turner syndrome and premature ovarian failure (POF). Semin Reprod Med 19:141, 2001 |
|
Boucher CA, Sargent CA, Ogata T, et al: Breakpoint analysis of Turner patients with partial Xp deletions: Implications for the lymphoedema gene location. J Med Genet 38:591, 2001 |
|
Bione S, Toniolo D: X chromosome genes and premature ovarian failure. Semin Reprod Med 18:51, 2000 |
|
Ford CE: Mosaics and chimaeras. Br Med Bull 25:104, 1969 |
|
Tharapel AT, Anderson KP, Simpson JL, et al: Deletion (X)(q26.1→q28) in a proband and her mother: Molecular characterization and phenotypic-karyotypic deductions Am J Hum Genet 52:463, 1993 |
|
Davison RM, Fox M, Conway GS: Mapping of the POF1 locus and identification of putative genes for premature ovarian failure. Mol Hum Reprod 6:314, 2000 |
|
Simpson JL: Genetics of female infertility. In Filicori M, Flamigni C (eds): Proceedings of the Conference, Treatment of Infertility: The New Frontiers. pp 37, 52 Boca Raton, FL, Communications Media for Education, 1998 |
|
Ogata T, Matsuo N: Turner syndrome and female sex chromosome aberrations: Deduction of the principal factors involved in the development of clinical features. Hum Genet 95:607, 1995 |
|
Wandstrat AE, Conroy JM, Zurcher VL, et al: Molecular and cytogenetic analysis of familial Xp deletions. Am J Med Genet 94:163, 2000 |
|
Thomas NS, Huson SM: Atypical phenotype in a female with a large Xp deletion. Am J Med Genet 104:81, 2001 |
|
Thomas NS, Sharp AJ, Browne CE, et al: Xp deletions associated with autism in three females. Hum Genet 104:43, 1999 |
|
Wilson MG, Modebe O, Towner JW, et al: Ullrich-Turner syndrome associated with interstitial deletion of Xp11.4 leads to p22.31 Am J Med Genet 14:567, 1983 |
|
Herva R, Kaluzewski B, de la Chapelle A: Inherited interstitial del(Xp) with minimal clinical consequences: With a note on the location of genes controlling phenotypic features. Am J Med Genet 3:43, 1979 |
|
Fraccaro M, Maraschio P, Pasquali F, et al: Women heterozygous for deficiency of the (p21 leads to pter) region of the X chromosome are fertile. Hum Genet 39:283, 1977 |
|
James RS, Dalton P, Gustashaw K, et al: Molecular characterization of isochromosomes of Xq. Ann Hum Genet 61:485, 1997 |
|
Massa G, Vanderschueren-Lodeweyckx M, Fryns JP: Deletion of the short arm of the X chromosome: a hereditary form of Turner syndrome. Eur J Pediatr 151:893, 1992 |
|
Knauff EA, Blauw HM, Pearson PL et al: Copy number variants on the X chromosome in women with primary ovarianinsufficiency. Fertil Steril. 2011 Apr;95(5):1584-8.e1. Epub 2011 Feb 12. |
|
Quilter CR, Karcanias AC, Bagga MR et al: Analysis of X chromosome genomic DNA sequence copy number variation associatedwith premature ovarian failure (POF). Hum Reprod. 2010 Aug;25(8):2139-50. Epub 2010 Jun 22. |
|
Dudding TE, Lawrence O, Winship I et al: Array comparative genomic hybridization for the detection of submicroscopic copynumber variations of the X chromosome in women with premature ovarian failure. Hum Reprod. 2010 Dec;25(12):3159-60; author reply 3160-1. Epub 2010 Oct 16. |
|
Aboura A, Dupas C, Tachdjian G et al: Array comparative genomic hybridization profiling analysis revealsdeoxyribonucleic acid copy number variations associated with premature ovarianfailure. J Clin Endocrinol Metab. 2009 Nov;94(11):4540-6. Epub 2009 Oct 16. |
|
Kok HS, van Asselt KM, van der Schouw YT et al: Genetic studies to identify genes underlying menopausal age. Hum Reprod Update. 2005 Sep-Oct;11(5):483-93. Epub 2005 Jul 15. |
|
Ochalski ME, Engle N, Wakim A et al: Complex X chromosome rearrangement delineated by array comparative genomehybridization in a woman with premature ovarian insufficiency. Fertil Steril. 2011 Jun;95(7):2433.e9-15. Epub 2011 Apr 29. |
|
Simpson JL, Elias S. Genetics in Obstetrics and Gynecology, 3rd edn. Philadelphia:WB Saunders, 2003 |
|
Simpson JL. Disorders of the gonads, genital tract, and genitalia. In: Principles and Practice of Medical Genetics, 5th edn. New York: Churchill-Livingstone, 2007:2005-92. |
|
Jones MH, Furlong RA, Burkin H et al: The Drosophila developmental gene fat facets has a human homologue in Xp11.4 Hum Mol Genet. 1996 Nov;5(11):1695-701. |
|
Luoh SW, Bain PA, Polakiewicz RD et al: Zfx mutation results in small animal size and reduced germ cell number in maleand female mice. Development. 1997 Jun;124(11):2275-84. |
|
Dube JL, Wang P, Elvin J et al: The bone morphogenetic protein 15 gene is X-linked and expressed in oocytes. Mol Endocrinol. 1998 Dec;12(12):1809-17. |
|
Di Pasquale E, Beck-Peccoz P, Persani L: Hypergonadotropic ovarian failure associated with an inherited mutation of human Am J Hum Genet. 2004 Jul;75(1):106-11. Epub 2004 May 10. |
|
Kovanci E, Heard MJ, McKenzie LJ et al. Mutation in the BMP15 gene is associated with premature ovarian failure. J Soc Gynecol Investig 2005; 12:236A |
|
Laissue P, Christin-Maitre S, Touraine P et al: Mutations and sequence variants in GDF9 and BMP15 in patients with prematureovarian failure. Eur J Endocrinol. 2006 May;154(5):739-44. |
|
Dixit H, Rao LK, Padmalatha VV et al: Missense mutations in the BMP15 gene are associated with ovarian failure. Hum Genet. 2006 May;119(4):408-15. Epub 2006 Mar 1. |
|
Ellison JW, Wardak Z, Young MF et al: PHOG, a candidate gene for involvement in the short stature of Turner syndrome. Hum Mol Genet. 1997 Aug;6(8):1341-7. |
|
Rao E, Weiss B, Fukami M et al: Pseudoautosomal deletions encompassing a novel homeobox gene cause growth failurein idiopathic short stature and Turner syndrome. Nat Genet. 1997 May;16(1):54-63. |
|
Belin V, Cusin V, Viot G et al: SHOX mutations in dyschondrosteosis (Leri-Weill syndrome). Nat Genet. 1998 May;19(1):67-9. |
|
Shears DJ, Vassal HJ, Goodman FR et al: Mutation and deletion of the pseudoautosomal gene SHOX cause Leri-Weilldyschondrosteosis. Nat Genet. 1998 May;19(1):70-3. |
|
Tachdjian G, Aboura A, Portnoi MF et al: Cryptic Xp duplication including the SHOX gene in a woman with 46,X,del(X)(q21.31) and premature ovarian failure. Hum Reprod. 2008 Jan;23(1):222-6. Epub 2007 Nov 1. |
|
Bione S, Sala C, Manzini C et al: A human homologue of the Drosophila melanogaster diaphanous gene is disrupted ina patient with premature ovarian failure: evidence for conserved function inoogenesis and implications for human sterility. Am J Hum Genet. 1998 Mar;62(3):533-41. |
|
Mumm S, Herrera L, Waeltz PW et al: X/autosomal translocations in the Xq critical region associated with prematureovarian failure fall within and outside genes. Genomics. 2001 Aug;76(1-3):30-6. |
|
Prueitt RL, Chen H, Barnes RI et al: Most X;autosome translocations associated with premature ovarian failure do not Cytogenet Genome Res. 2002;97(1-2):32-8. |
|
Lacombe A, Lee H, Zahed L et al: Disruption of POF1B binding to nonmuscle actin filaments is associated withpremature ovarian failure. Am J Hum Genet. 2006 Jul;79(1):113-9. Epub 2006 May 26. |
|
Bione S, Rizzolio F, Sala C et al: Mutation analysis of two candidate genes for premature ovarian failure, DACH2 andPOF1B. Hum Reprod. 2004 Dec;19(12):2759-66. Epub 2004 Sep 30. |
|
Bione S, Toniolo D: X chromosome genes and premature ovarian failure. Semin Reprod Med. 2000;18(1):51-7. |
|
Prueitt RL, Ross JL, Zinn AR: Physical mapping of nine Xq translocation breakpoints and identification of XPNPEP2 as a premature ovarian failure candidate gene. Cytogenet Cell Genet 89:44, 2000 |
|
Mansouri MR, Schuster J, Badhai J et al: Alterations in the expression, structure and function of progesterone receptor Hum Mol Genet. 2008 Dec 1;17(23):3776-83. Epub 2008 Sep 9. |
|
Katsuya T, Horiuchi M, Minami S et al: Genomic organization and polymorphism of human angiotensin II type 2 receptor: noevidence for its gene mutation in two families of human premature ovarian failuresyndrome. Mol Cell Endocrinol. 1997 Mar 28;127(2):221-8. |
|
Wittenberger MD, Hagerman RJ, Sherman SL et al: The FMR1 premutation and reproduction. Fertil Steril. 2007 Mar;87(3):456-65. Epub 2006 Oct 30. |
|
Schwartz CE, Dean J, Howard-Peebles PN et al: Obstetrical and gynecological complications in fragile X carriers: a multicenterstudy. Am J Med Genet. 1994 Jul 15;51(4):400-2. |
|
Allingham-Hawkins DJ, Babul-Hirji R, Chitayat D et al: Fragile X premutation is a significant risk factor for premature ovarian failure: Am J Med Genet. 1999 Apr 2;83(4):322-5. |
|
Sullivan AK, Marcus M, Epstein MP et al: Association of FMR1 repeat size with ovarian dysfunction. Hum Reprod. 2005 Feb;20(2):402-12. Epub 2004 Dec 17. |
|
Foresta C, Ferlin A, Gianaroli L et al: Guidelines for the appropriate use of genetic tests in infertile couples. Eur J Hum Genet. 2002 May;10(5):303-12. |
|
Yachelevich N, Gittler JK, Klugman S et al: Terminal deletions of the long arm of chromosome X that include the FMR1 gene infemale patients: a case series. Am J Med Genet A. 2011 Apr;155A(4):870-4. doi: 10.1002/ajmg.a.33936. Epub 2011 |