Pediatric Ovarian Malignancies
Authors
INTRODUCTION
Ovarian malignancies in children may represent an array of unique problems for the clinician who is more accustomed to diagnosing and treating ovarian neoplasia in adults. Although ovarian malignancies in children are rare (representing only 0.2% of all ovarian neoplasms), their recognition and diagnosis are vital because they can be fulminant if treated inadequately. Advances in combination chemotherapy have enabled patients to survive the disease with tolerable (less toxic) short-term and long-term morbidity. Whenever possible, treatment should be individualized to preserve reproductive and menstrual function without jeopardizing the life of the child.
Tumors of germ cell origin (e.g., mature teratoma, malignant teratoma, endodermal sinus tumor [yolk sac tumor], embryonal carcinoma, dysgerminoma, primary choriocarcinoma) constitute approximately 70% of ovarian tumors in children (Table 1).1 All such tumors with the exception of the mature teratoma are malignant. The dysgerminoma in its pure form is considered a low-grade malignancy. Because these tumors have a common histogenesis, mixed germ cell tumors can be found, which yield a confusing clinical picture with regard to anticipated post-therapy responses.
TABLE 1. Incidence of Ovarian Malignancies in 874 Children According to Histologic Tumor Types
Diagnosis | N | Malignant Tumors (%) |
Germ Cell | ||
Dysgerminoma | 214 | 24.5 |
Immature teratoma | 172 | 19.7 |
Mixed germ cell | 73 | 8.4 |
Endodermal sinus | 120 | 13.7 |
Embryonal | 25 | 2.9 |
Primary choriocarcinoma | 8 | 0.9 |
Total | 612 | 70.1 |
Sex-Cord Stromal | ||
Pure granulosa cell | 46 | 5.3 |
Juvenile granulosa cell | 30 | 3.4 |
Granulosa-theca cell | 21 | 2.4 |
Sertoli-Leydig cell | 23 | 2.6 |
Fibrosarcoma | 5 | 0.6 |
Sarcoma | 3 | 0.3 |
Pure Sertoli cell | 3 | 0.3 |
Nonspecific | 17 | 1.9 |
Total | 148 | 16.8 |
Surface Epithelial-Stromal Tumors | ||
Serous cystadenocarcinoma | 23 | 2.6 |
Mucinous cystadenocarcinoma | 20 | 2.3 |
Clear cell adenocarcinoma | 8 | 0.9 |
Endometrioid adenocarcinoma | 2 | 0.2 |
Adenocarcinoma | 13 | 1.5 |
Total | 66 | 7.5 |
Metastatic and Unclassified | ||
Lymphoma | 19 | 2.2 |
Adrenal rest | 1 | 0.1 |
Metastatic | 3 | 0.3 |
Unclassified | 25 | 2.9 |
Total | 48 | 5.5 |
TOTAL | 874 |
Surface epithelial stromal tumors (common epithelial tumors) are rare in children and represent the same morbidity and mortality in children as in adults; therapy also is similar to that used in adults. Granulosa-stromal cell tumors are the most common of the sex-cord stromal cell tumors, vary greatly in their clinical course, and are rare in children. This chapter reviews the world literature on the clinical characteristics and modern management of the aforementioned tumors, using the new histologic classification of ovarian tumors developed by the World Health Organization (Table 2).
TABLE 2. Histologic Classification of Ovarian Tumors*

INCIDENCE
The average overall incidence of ovarian neoplasms is 1.7:100,000 per year.2,3 The rarity of ovarian tumors in children precludes a statistically significant compilation of the age-related occurrence of specific tumors. Most data are reported from referral centers, where distorted patient sampling results in a wide range of statistics. Despite these variations, however, certain age-related trends concerning the incidence and histology of pediatric ovarian tumors are evident. Germ cell tumors predominate in all age groups, accounting for approximately 70% of ovarian neoplasms in children and adolescents (this is in marked contrast to the 20% incidence of similar tumors in adults).3
Any ovarian enlargement is rare in infants. Most enlargements indicate the presence of a non-neoplastic follicle or theca-lutein cysts.4 In a review of 14 large series, 17% of tumors were found in children between birth and 4 years of age.5 In infants, tumors of germ cell and sex-cord stromal origin constitute 5% and 10% of ovarian lesions.6 With advancing age, sex-cord stromal and surface epithelial-stromal tumors increase in frequency, particularly after menarche. In the 10- to 14-year-old group, sex-cord stromal tumors constitute 5% to 10% and surface epithelial-stromal tumors constitute approximately 12% to 25% of ovarian neoplasms. After puberty, the balance continues to shift toward a preponderance of surface epithelial-stromal tumors, which, if all ages are considered, constitute 70% of ovarian tumors.1
In 18 studies published from 1940 to 1965, of 464 pediatric ovarian neoplasms, an average of 22% were malignant and 78% were benign. In 15 subsequent studies published from 1966 to 1985, the incidence of malignancy ranged from 13% to 85%. In seven studies published from 1986 to 1993, of 456 neoplasms, 29% were malignant and 71% were benign. Of a total of 2026 childhood ovarian tumors reported in studies published from 1940 to 1993, 33% were malignant and 67% were benign (Table 3).7,8,9,10,11,12,13,14,15,16,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34
TABLE 3. Literature Review: Ovarian Tumors in Children, 1940–1993—Malignant Versus Benign
Author | Year | Total No. of Tumors | No. Malignant | No. Benign |
Gross | 1940 | 25 | 6 | 19 |
Costin | 1949 | 22 | 2 | 20 |
Schaefer | 1949 | 6 | 2 | 4 |
Chisholm | 1949 | 8 | 2 | 6 |
Gagner | 1949 | 39 | 15 | 24 |
Butt | 1955 | 23 | 3 | 20 |
Carache | 1959 | 6 | 5 | 1 |
Forshall | 1959 | 21 | 6 | 15 |
Darte | 1960 | 41 | 6 | 35 |
Radman | 1960 | 27 | 3 | 24 |
Dargem | 1960 | 19 | 10 | 9 |
Adams | 1961 | 13 | 2 | 11 |
Boler | 1961 | 16 | 1 | 15 |
Reis | 1962 | 25 | 1 | 24 |
Garfunkel | 1962 | 13 | 0 | 13 |
Thatcher | 1963 | 21 | 8 | 13 |
Groeber | 1963 | 13 | 2 | 11 |
Abell | 1965 | 188 | 51 | 137 |
Thompson | 1967 | 41 | 24 | 17 |
Moore | 1967 | 83 | 11 | 72 |
Kilman | 1967 | 18 | 6 | 12 |
Ein | 1970 | 49 | 12 | 37 |
Norris | 1972 | 353 | 186 | 167 |
Acosta | 1972 | 34 | 8 | 26 |
Sawai | 1973 | 67 | 57 | 10 |
Harris | 1974 | 26 | 6 | 20 |
Towne | 1975 | 55 | 16 | 39 |
Adelman | 1975 | 43 | 8 | 35 |
Giugiaro | 1975 | 14 | 3 | 11 |
Yakushiji | 1981 | 76 | 40 | 36 |
Golladay | 1983 | 16 | 6 | 10 |
Ehren | 1984 | 63 | 16 | 47 |
Shawis | 1985 | 106 | 12 | 94 |
Cronen | 1988 | 30 | 8 | 22 |
Khanna | 1988 | 135 | 46 | 87 |
Schuck | 1990 | 47 | 6 | 41 |
Haefner | 1992 | 101 | 32 | 68 |
Warner | 1992 | 23 | 10 | 13 |
Skinner | 1993 | 29 | 12 | 17 |
Brown | 1993 | 91 | 19 | 72 |
TOTAL | 2026 | 669 (33%) | 1357 (67%) |
CLINICAL PRESENTATION
The variety of symptoms associated with an enlarging ovarian lesion in pediatric patients commonly obscures the cause of the complaint and unduly delays diagnosis and treatment. Initial symptoms may be absent or vague, and the average delay between clinical onset and histologic diagnosis is 3 to 4 months.35 Symptoms relate to the tumor’s rate of growth, location, histologic type, and degree of malignancy and whether or not there is metastasis, endocrine activity, and associated complications (e.g., torsion, rupture, infection, or hemorrhage). The spectrum of symptoms referable to ovarian neoplasms in children is extensive.
The most common presenting symptom is abdominal pain, which is present in more than half of patients.10,11 In the prepubertal child, the pain is classically periumbilical, owing to the abdominal location of the ovary and the limited confines of the bony pelvis, which permits only upward expansion of the tumor. In one series, 80% of children younger than 10 years old had appreciable abdominal swelling, whereas only 32% of patients older than 12 years exhibited similar findings.7
Although pelvic pain can result from peritoneal stretching, pressure on adjacent organs, rupture, or hemorrhage into a cyst, the most common cause is torsion. The long suspensory ligament of the ovary predisposes the child to volvulus (incidence range, 14% to 71%). Of mature teratomas, 30% are detected by acute onset of abdominal pain, often accompanied by nausea; vomiting; and radiation of pain to the groin, lower extremity, or costovertebral areas—all of which are subsequent to torsion.10,11,23
Isosexual precocious puberty is associated with ovarian tumors in 5% of patients; however, approximately 16% of children with ovarian tumors present with premature secondary sexual development. In a compilation of 43 cases of tumor-related precocious puberty, the pathologic diagnosis included granulosa cell tumor (28%), granulosa-theca cell tumor (14%), immature teratoma (14%), adenocarcinoma (7%), and Sertoli cell tumor (5%). In 25% of cases, the only identifiable lesions were non-neoplastic cysts of follicular origin. In patients with detectable endocrine disturbances, the ovarian lesion usually is palpated as either an abdominal or a pelvic mass. The malignancy rate for endocrinologically functioning ovarian tumors is 50% to 60%.36
Germ cell tumors have been associated with pregnancy in a small percentage of cases. Dysgerminomas are the most common germ cell tumor diagnosed during pregnancy or in the immediate postpartum period.37
DIAGNOSIS
The rarity of pediatric ovarian malignancies contributes to a low index of suspicion: Only 36% to 63% of cases are identified correctly before surgery. On vaginal, abdominal, or rectal examination, a pelvic or abdominal mass may be detected in 88% of patients. A palpable ovary in the prepubertal patient is presumed to be abnormal because of the absence of gonadotropins. The most common erroneous diagnosis is acute appendicitis; 2% of patients given this diagnosis are found to have an ovarian tumor with volvulus.38 Although surgical resection and histologic examination are the only definitive means of diagnosing an ovarian neoplasm, several radiographic, ultrasonographic, and laboratory parameters can aid the clinician.
Radiography
Radiographic assessment of the abdomen may show soft tissue densities suggestive of ascites, intestinal obstruction, or tumor calcification. A chest radiograph may show a pleural effusion consistent with Demons-Meigs syndrome or pulmonary metastasis. Intravenous pyelography and barium enema rule out intrinsic urinary or gastrointestinal tract pathology.
Ultrasonography
Ultrasonography provides information regarding the size, density, character, and location of the tumor.19 Computed tomography and magnetic resonance imaging may afford additional visualization of the abdomen and pelvis and indicate lymph node and retroperitoneal involvement.
Laboratory Parameters
In addition to routine blood chemistry studies and urinalysis, several tumor marker assays are helpful. Alpha-fetoprotein (AFP) levels can be elevated in embryonal carcinomas, yolk sac tumors, and teratoid tumors with a significant yolk sac tumor component. β-Human chorionic gonadotropin (β-hCG) is present in nongestational choriocarcinomas and may be minimally elevated in the dysgerminoma variant with syncytiotrophoblast cells. AFP and β-hCG levels are elevated in embryonal carcinomas. The monoclonal antibody CA-125 may help determine therapeutic responses and may detect recurrences of surface epithelial stromal tumors, especially those of the serous variety. Inhibin levels may be elevated in mucinous and granulosa cell tumors. Measurement of carcinoembryonic antigen may reveal increased levels in ovarian malignancies, but the measurement is of little value as a diagnostic test because it lacks specificity. Bone age determinations, vaginal smears, and hormonal assays are helpful in assessing premature development of secondary sex characteristics.
GERM CELL TUMORS
Germ cell tumors are the most common ovarian neoplasms appearing in the first two decades of life. These tumors decrease in incidence with advancing age. In adults, germ cell tumors constitute only 10% to 15% of ovarian tumors. The degree of malignancy also decreases as the ovary matures. The more malignant varieties (i.e., embryonal and yolk sac tumors) are most common in very young patients. Mixed germ cell tumors, mature teratomas, and dysgerminomas occur at a later age; for example, mature teratomas are the most prevalent during the second decade of life.
DYSGERMINOMAS
Dysgerminomas are the most common ovarian malignancies in children, constituting 9.5% to 11% of childhood ovarian tumors and 24.5% of pediatric ovarian malignancies.10 There is an increased frequency of dysgerminomas among patients with genetically abnormal gonads. Dysgerminomas have been detected in 7-month-old infants; 7% of dysgerminomas are found in patients younger than 10 years, and 34% are found in the 10- to 19-year-old group. The average age at the time of detection is 22 years.
A pure dysgerminoma is endocrinologically inactive. Signs of pronounced hormonal activity indicate the presence of a functioning component, placing the tumor into a mixed germ cell category. A minimally elevated β-hCG may herald the existence of a dysgerminoma variant with syncytioblast cells.
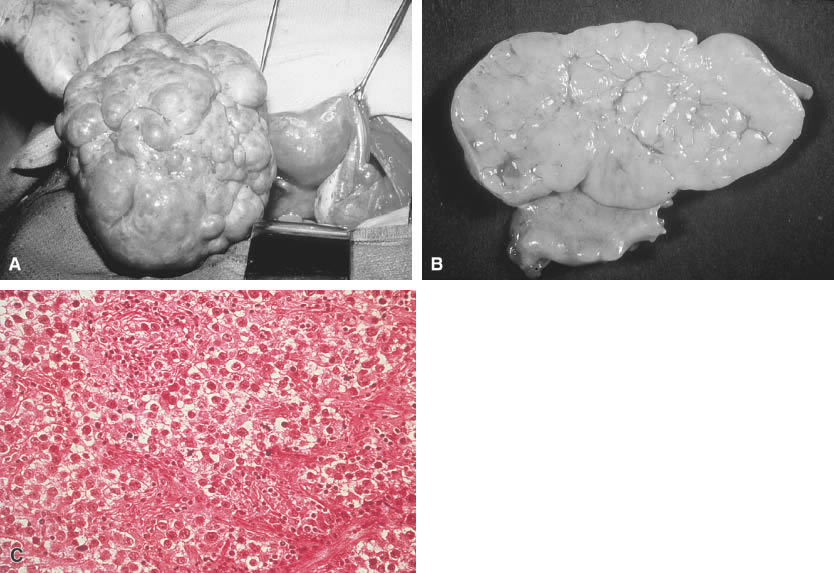
Grossly, dysgerminomas are rubbery, gray, smooth, or bosselated and are surrounded by a dense capsule (Fig. 1A). The cut surface is soft and homogeneous and has a brainlike consistency (Fig. 1B). Tumors range in diameter from several centimeters to 50 cm. The rate of bilateralism is reported to be 5% to 20%.
{kind=link}
The histologic picture of dysgerminomas is analogous to that of undifferentiated germ cells of the embryonal gonad. These well-defined clusters of cells are separated by fibrous tissue septa with lymphocytic infiltration and an occasional Langhans’ giant cell (Fig. 1C). Although no single pathologic factor is related directly to prognosis, a marked inflammatory response suggests a good immunologic reaction in the tumor. Marked cellular atypia, frequent mitosis, and mixtures with more lethal germ cell tumors yield a poorer prognosis.
The malignancy of pure dysgerminomas is relatively low grade, with survival rates of 80% to 96%. The incidence of recurrence is reportedly high, however—33% to 50% in some studies.24 Although most recurrences are seen within the first 3 years after therapy, later recurrences have been reported. Predisposing factors for recurrence of dysgerminomas include a history of no adjunctive treatment and peritoneal spillage, capsule extension, bilaterality, and a mixture of other malignant germ cell tumors.24
Treatment
In the past, because there is a 10% to 15% incidence of bilaterality and lymphatic metastases, a wedge biopsy of the contralateral ovary and ipsilateral pelvic para-aortic lymphadenectomy were mandatory procedures. With the advent of newer chemotherapy regimens and the exquisite chemosensitivity of dysgerminomas, mandatory contralateral wedge biopsy and ipsilateral lymphadenectomy have been called into question in an attempt to limit iatrogenic reproductive compromise.
Salpingo-oophorectomy is used to treat pediatric unilateral dysgerminomas. If the lesion is a stage IA (negative peritoneal washings, lymph nodes, and omental biopsies; no capsular penetration, ascites, or detectable metastases), no postoperative therapy is indicated. If the dysgerminoma involves both ovaries, bilateral salpingo-oophorectomy and a full staging operation should be carried out; however, unless the uterus is involved by cancer, it should not be removed because in vitro fertilization with a donor ovum can be an option for the patient. Metastatic disease demands complete cytoreductive surgery. Recurrent disease can be surgically resected and subsequently treated with chemotherapy or radiotherapy. Of recurrences, 75% appear within 1 year of initial surgery and 80% appear within 2 years. The abdomen and pelvis are the most frequent sites of surgical failure, followed by the para-aortic and supraclavicular nodes.40
In the past, many stage I patients and all patients of higher stage were treated with radiotherapy because dysgerminomas have an inherently high degree of radiosensitivity. Pelvic radiotherapy is associated with a high incidence of sterility, however; with the development of effective chemotherapy regimens, radiotherapy is rarely indicated except for unusual metastatic patterns.
Before the mid-1980s, the chemotherapy experience in early and advanced dysgerminomas was anecdotal. In 1984, dysgerminoma patients became eligible for Gynecologic Oncology Group (GOG) chemotherapy protocols for the treatment of advanced ovarian germ cell tumors. In the first protocol, patients received a cis-platinum, vinblastine, bleomycin (PVB) regimen. The second and ongoing study involves induction chemotherapy with bleomycin, etoposide, and cis-platinum (BEP), followed by consolidation with vincristine, actinomycin D, and cyclophosphamide (VAC). In his review article, Williams41 reported that 19 of 20 patients with stage III or IV disease were found to be disease-free at follow-up (range, 9 to 66 months; median, 26 months). Williams41 stated:
From this data, it would seem that nearly all patients with advanced dysgerminoma will be durable complete responders after treatment with platinum-based chemotherapy. In addition, preliminary evidence suggests that chemotherapy-treated patients have a reasonably high likelihood of resuming menses and recovering fertility. Adjuvant chemotherapy will almost always prevent relapse in patients with germ cell tumors other than dysgerminoma; data are limited in dysgerminoma but are equally favorable. Thus, patients who have an intact contralateral ovary, tube, and uterus, who are desirous of bearing children and who are deemed to require adjuvant therapy should be treated with chemotherapy and not radiation. Because of the effectiveness and generally low toxicity, it is likely that similar therapy is also appropriate for patients in whom the fertility is moot.
The current recommendation for adjuvant chemotherapy in the treatment of metastatic germ cell tumors is BEP.42,43 This regimen has shown a sustained remission rate of 75% and surpassed the previously recommended VAC therapy.43
IMMATURE TERATOMAS
Immature teratomas constitute 28% of germ cell tumors and 19.7% of ovarian malignancies in children. This neoplasm is extremely rare in adults. It is found on average at age 11 years, although cases have been reported in patients as young as 14 months old.41
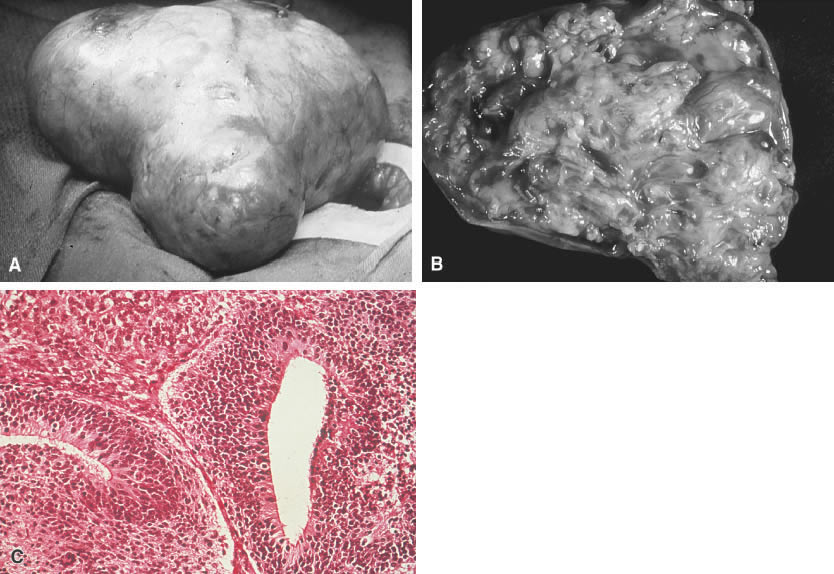
The immature teratoma, also referred to as an embryonal teratoma, teratocarcinoma, solid teratoma, malignant dermoid, embryoma, and teratoblastoma, may be grossly similar to its benign counterpart, the mature teratoma. These tumors range in size from 7 to 28 cm and are encapsulated, bosselated, and usually solid with cystic areas (Fig. 2A and B). Bilateral involvement is seen in approximately 10% of cases.
{kind=link}
Histologically the immature teratoma exhibits a mixture of mature and immature tissue forms of mesenchymal and epithelial origin, reminiscent of the developing stages of the embryonic tissues (Fig. 2C). The most common pattern seems to be an undifferentiated cellular stroma with structures suggesting immature neural epithelium. Fetal glands and squamous epithelium are seen with mesenchymal derivatives, such as cartilage, bone, smooth muscle, neuroglia, and ganglion cells. Formations suggestive of other germ cell tumors may be identified.
The immature teratoma is not to be confused with a mature teratoma in which one tissue element has undergone malignant transformation. The latter is focal, is found incidentally in elderly patients with long-standing tumor (usually squamous cell carcinoma), and is associated with a more favorable prognosis.
The histologic grade of the primary tumor and its clinical stage are the main determinants of extra-ovarian spread. When metastasis has occurred, the histologic grade of the metastatic lesion is the major factor in predicting prognosis.45 Immature teratomas are graded on the basis of the amount of cellular immaturity. The grading scheme ranges from grade 0 (a solid teratoma with all tissue mature) to grade 3 (markedly atypical immature embryonal tissue with a high degree of mitotic activity).
Treatment
Biopsy and grading must be done for all peritoneal metastases. After oophorectomy, stage I (limited to the ovary)/grade 1 patients should be followed conservatively. All other patients who have evidence of extraovarian spread, if the tumor is grade 2 or 3 or if its implants or recurrences are graded 1, 2, or 3 (even if stage IA), should receive adjuvant triple chemotherapy.43,46 PVB has been used successfully to treat patients who have had VAC failures.47 Preliminary results from a GOG trial indicate that 50 of 52 stage I, II, and III patients with completely resected disease are disease-free after three cycles of BEP.48 It has been suggested that a predominance of malignant neural tissue or thyroid tissue (struma ovarii) indicates a more optimistic prognosis than the finding of chorioepitheliomatous or vitelline elements. BEP chemotherapy, in 5-day regimens, is currently the treatment of choice because of the decreased toxicity of etoposide compared with vinblastine (i.e., in PVB treatment, which apparently is superior to VAC chemotherapy).3,16,43,49
Despite the fact that 70% of immature teratomas are theoretically stage IA, the overall mortality rate was greater than 75% until the advent of chemotherapy. A marked propensity for rapid growth and metastasis is seen even in early stage IA lesions. Because the 5-year survival rate is unaffected by the extent of surgery in stage IA lesions, the surgical treatment of choice is a unilateral salpingo-oophorectomy. Removal of the uterus, including uterine tubes and ovaries, and the omentum is recommended if there is local extension, metastasis, or contralateral involvement. Radiotherapy offers no benefit.
YOLK SAC (ENDODERMAL SINUS) TUMORS
Yolk sac tumors constitute 6% of pediatric ovarian tumors and 7.2% of ovarian malignancies in children. The average age of patients at diagnosis is 13 to 14 years, but tumors reportedly have been found in patients 7 months old.35
Controversy exists concerning the histopathology of yolk sac tumor (also named endodermal sinus tumor, embryonal carcinoma, mesonephroma ovarii of Schiller, Teilum tumor, extraembryonic mesoblastoma, yolk sac carcinoma, and mesometanephric rest tumor), and numerous theories have arisen to explain its histogenesis. Elevated serum AFP levels in these tumor cells show their close relationship with the yolk sac, which is the source of AFP in the human fetus.
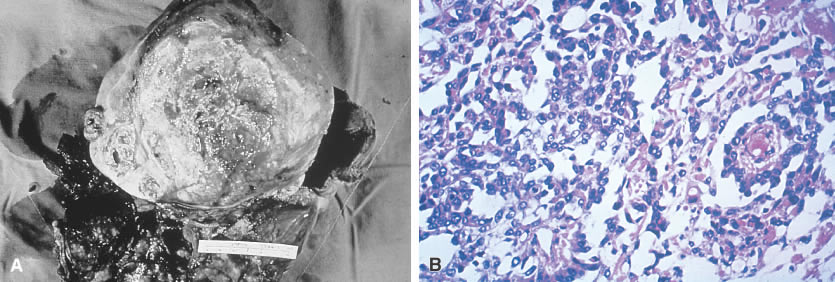
The gross appearance of the yolk sac tumor is classically yellow-red-gray tissue that is soft and friable, with a thin, smooth capsule. Cystic spaces are common, and areas of hemorrhage and necrosis occasionally are found (Fig. 3A). The neoplasm grows rapidly and may be densely adherent to surrounding tissues.
{kind=link}
Microscopically the yolk sac tumor exhibits a fairly constant pattern of a loose network of pleomorphic, poorly differentiated cells. These cells have an epithelial appearance in some areas, with papillary clusters thrusting into cystic spaces (Fig. 3B). Extracellular and intracellular round or oval hyaline globules (Schiller-Duval bodies) are a frequent finding.
Treatment
Yolk sac tumors are highly malignant and previously were almost uniformly fatal before chemotherapy. The rapid growth of this tumor precludes conservative surgical approaches: Widespread metastasis usually is found on initial celiotomy. The previous 5-year mortality rate of nearly 100% is not improved with extensive surgery alone; most patients died within 6 to 12 months if they were not treated with chemotherapy. Radiotherapy is ineffectual. The general consensus is that all patients, regardless of stage, should receive chemotherapy for this type of tumor. Chemotherapy with VAC or PVB has made an impressive impact on the clinical course of this disease, especially after optimal debulking. Disease-free intervals range from 2 to 8 years.44,48,49,50 Reports indicate that PVB and BEP are at least as effective and possibly better than VAC. Although no randomized trials exist comparing VAC with PVB or BEP, it is likely that the latter two regimens are preferable therapy for yolk sac tumor.43,49,53 Now, with aggressive chemotherapy, an 80% overall survival is achievable.54 Therapeutic responses to chemotherapy and recurrences can be monitored by serial determination of AFP.
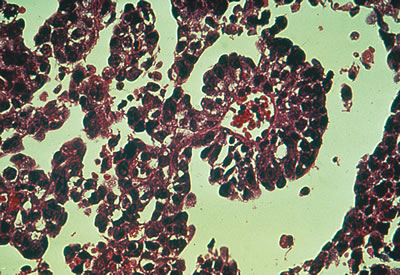
Considerable controversy has surrounded embryonal carcinoma, a rare germ cell tumor often confused with yolk sac tumor. This entity constitutes less than 3% of malignant ovarian germ cell tumors and resembles the embryonal carcinoma seen in adult testicles (Fig. 4). It is characterized by elevations of β-hCG and AFP. The extreme rarity of this lesion renders significant statistical evaluation impossible. The prognosis and treatment are similar to those of the yolk sac tumor.
{kind=link}
PRIMARY CHORIOCARCINOMA
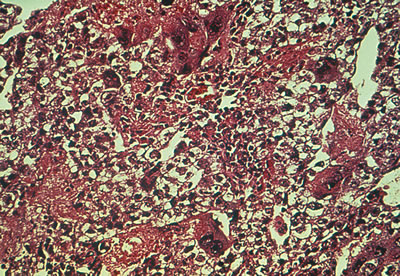
Primary choriocarcinomas of the ovary are extremely rare (0.6% of germ cell tumors and 0.9% of ovarian malignancies in children). A true primary choriocarcinoma is believed to arise from the primordial germ cell. This tumor also may form in a teratoma of the ovary or may metastasize to the ovary from an extragonadal teratoma. In ovarian choriocarcinoma occurring after puberty, gestational choriocarcinoma must be ruled out because most trophoblastic neoplasms of the ovary arise from ovarian pregnancies or represent metastasis from an unrecognized uterine or tubal gestation. Choriocarcinoma, primary or gestational, produces β-hCG, which can be detected in blood and urine.
On gross examination, primary choriocarcinomas are hemorrhagic and friable. Microscopically, they consist of cells resembling syncytiotrophoblasts and cytotrophoblasts, with extensive areas of hemorrhage and necrosis (Fig. 5).
{kind=link}
Treatment
Primary choriocarcinoma in children is rapidly fatal; no patient has been reported to survive more than 8 months after diagnosis. Rapid hematogenous metastasis and local extension are characteristic of this neoplasm in children. Symptoms and organ involvement in children are similar to findings in menstruating women with gestational choriocarcinoma. Radical surgery offers no benefit over conservative extirpation of the involved gonad. The tumor is radioresistant and historically rarely responds to chemotherapeutic agents employed so successfully against its gestational counterpart. Occasionally, success has been reported with a triple regimen including actinomycin D, methotrexate, and chlorambucil.
MIXED GERM CELL TUMOR
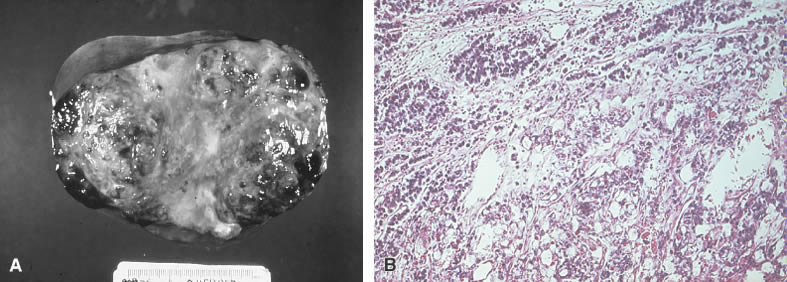
Neoplasms containing two or more germ cell derivatives account for 4% of ovarian tumors and 8.6% of ovarian malignancies in children or adolescents. The yolk sac tumor is seen in 88% of mixed germ cell tumors; choriocarcinoma, the second most frequent germ cell element, is seen in 25%. These aggressive components are found most often in conjunction with dysgerminoma (Fig. 6).
| |
{kind=link}
Treatment
The biologic behavior of mixed germ cell tumors follows that of its most malignant component. The prognosis is generally poor because of the preponderance of a yolk sac or choriocarcinomatous component. Most of these tumors result in death in less than 1 year, despite treatment with VAC chemotherapy.44,50 Given the reported success of BEP in dysgerminomas, immature teratomas, and yolk sac tumors, this combination may be recommended as the treatment of choice.43
SEX-CORD STROMAL CELL TUMORS
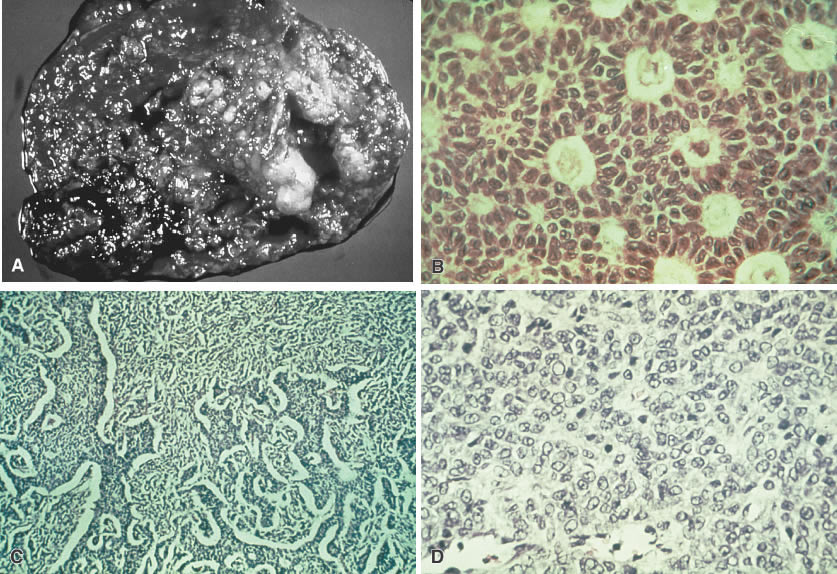
Sex-cord stromal cell tumors are less common than germ cell neoplasms in the first two decades of life. These tumors are generally of low-grade malignancy and comprise 12.6% of all pediatric ovarian neoplasms and 17% of ovarian malignancies in children. The primary sex-cord stromal cell tumors are the granulosa-stromal cell tumors and the Sertoli-stromal cell tumors (the gynandroblastoma and the pure Leydig cell tumor have not been reported in pediatric patients) (Fig. 7).
{kind=link}
The primary significance of neoplasms of mesenchymal origin is their capacity to be endocrinologically active. The granulosa-stromal cell tumors, which represent half of the potentially malignant sex-cord stromal tumors, are the most common neoplasms causing gonadal isosexual precocious puberty. Most cases of isosexual precocity are idiopathic, however, and less than 5% are associated with granulosa-stromal cell tumors. The postpubertal child may present with hypermenorrhea (75%) or amenorrhea (25%) as a result of the estrogen secretion from the granulosa-stromal cell tumor.55
Heterosexual precocious puberty may be stimulated by the most common masculinizing tumor of the ovary, androblastoma (arrhenoblastoma or Sertoli-Leydig cell tumor), but only six prepubertal cases have been reported. If the Sertoli cells predominate, the effects of excess estrogen may be seen; in a tumor composed primarily of Leydig cells, the androgenic effect prevails.
Juvenile granulosa cell tumors have been described.17,26,56 These tumors have a characteristic appearance of cellular nodules within a myxoid spindle cell stroma, and larger cells in a predominantly diffuse or solid pattern with vitalization of its cellular components. Of granulosa-stromal cell tumors in children younger than 16 years, 80% were found to fit this description. Two thirds of children with these tumors present with endocrine abnormalities, such as precocious pseudopuberty, virilization, or menstrual irregularities. The juvenile granulosa cell tumors are usually unilateral, present as stage IA, and seldom recur.
The prognosis for the sex-cord stromal tumors is generally excellent. Because there are no reliable histologic criteria to differentiate between benign and malignant lesions, malignant tumors are confirmed only by recurrence, local extension, or, less frequently, metastasis. Between 5% and 30% of granulosa-stromal cell tumors and fewer than 15% of androblastomas are malignant. The granulosa-stromal cell tumors have a propensity for late recurrence; in one instance, recurrence occurred 33 years after treatment. The androblastomas show a lesser malignant potential, with an 85% to 95% survival rate noted.
Treatment
Unilateral salpingo-oophorectomy is the treatment of choice for sex-cord stromal tumors. Because the incidence of bilateralism is low (granulosa-stromal cell, 2.5%; androblastoma, 4%), a wedge resection or bivalving of the contralateral ovary is not indicated if the gonad appears grossly normal. Radiotherapy is useful for metastatic disease or tumor recurrence, particularly in granulosa cell tumors. PVB and more recently BEP chemotherapy has been advocated.43,55,56,57,58,59
SURFACE EPITHELIAL-STROMAL TUMORS
The surface epithelial-stromal tumor, representing 80% to 90% of adult ovarian neoplasms, account for only 7% of malignancies in children.15 Endometrial and clear cell carcinomas are not found in the first two decades of life.23,37 The mucinous and serous cystadenocarcinomas are rare before puberty and have not been reported in children younger than 4 years old.13 The incidence of these coelomic epithelial lesions increases with advancing age.
The gross and microscopic characteristics of these tumors in adolescents are identical to those of similar neoplasms in adults. The tumors are bilateral in 10% of cases compared with 25% in adults.
Treatment
Although the malignant potential of surface epithelial-stromal tumors in children (7.1% to 13.5% of cystadenomas) is less than is seen in adults, malignant neoplasms in children show a clinical course and mortality similar to what is seen in adults. Serous lesions have the poorest prognosis. Tumors of low malignant potential behave in a generally benign fashion and, if properly staged as IA, may be treated with salpingo-oophorectomy only.
For malignant surface epithelial-stromal tumors other than stage IA, treatment includes total abdominal hysterectomy, bilateral salpingo-oophorectomy, omentectomy, pelvic/para-aortic lymph node sampling, and appendectomy. Chemotherapy regimens of cis-platinum or carboplatin with an alkylating agent (e.g., cyclophosphamide [Cytoxan] or PVB) generally are indicated for adjunctive therapy.2,17,56 Radiotherapy with a moving strip technique may be beneficial. The 10-year actuarial survival rate for all stages is 75% primarily because of the preponderance of early-stage lesions.17 Paclitaxel (Taxol) has shown promise in cis-platinum-resistant patients and may replace cyclophosphamide as primary therapy.60
GENERAL PRINCIPLES OF THERAPY
Gonadal tumors in children carry an overall malignancy rate of 35%. This fact must be balanced with the desire to maintain the child’s reproductive and developmental potential. A definitive histologic diagnosis and a thorough understanding of the behavior of each of these tumors are necessary for appropriate therapy. Initial conservatism is essential in the surgical treatment of pediatric ovarian tumors. A diagnosis from a frozen tissue section should not be used as the basis for radical surgery for a unilateral tumor in an otherwise unremarkable pelvis. Peritoneal washings and diaphragmatic scrapings for cytology with a radioisotopic and radiographic oncology survey dictate whether further therapy is necessary. A second surgical procedure may be required, depending on the tumor type and histologic grade.
The approach to extend surgical staging in children is controversial. Because of the low incidence of bilateralism and lymphatic metastasis in most pediatric ovarian tumors, a wedge biopsy of the contralateral ovary and pelvic/para-aortic lymphadenectomy are rarely indicated. In stage IA lesions, dysgerminomas (with their characteristic bosselated appearance and increased frequency of bilaterality and lymphatic metastasis) may be the one exception, although this is debated because of their exquisite chemosensitivity. Unilateral salpingo-oophorectomy provides adequate surgical treatment for tumors of low-grade malignancy (e.g., dysgerminomas granulosa-stromal cell tumors, androblastomas, and cystadenocarcinomas of low malignant potential). Unilateral salpingo-oophorectomy is also sufficient in tumors of a greater malignant nature, if staged as IA, in which case additional surgery does not improve the prognosis. This category includes immature teratomas, yolk-sac tumors, embryonal carcinomas, and nongestational choriocarcinomas, in which adjunctive therapy is mandatory. Total abdominal hysterectomy, bilateral salpingo-oophorectomy, omentectomy, and appendectomy are indicated in germ cell and sex-cord stromal cell tumors staged beyond IA. Because of the poor prognosis in dedifferentiated serous and mucinous cystadenocarcinomas, even when stage IA, the reproductive organs should be extirpated. Mucinous and serous cystadenocarcinomas with a low malignant potential (borderline) are less virulent and, when properly staged as IA, can be handled conservatively with a unilateral salpingo-oophorectomy.
There is some disagreement regarding the optimal treatment of patients with granulosa-stromal cell tumors or dysgerminomas who received conservative surgical therapy in childhood and who have completed their desired childbearing. Exploratory laparotomy with a hysterectomy and salpingo-oophorectomy has been suggested for these patients. Because of the theoretical potential for late recurrences the rarity of these tumors, and the unknown benefit of this surgical intervention, however, most clinicians do not recommend this method of treatment.
REFERENCES
Benson R: Ovarian tumors in childhood and adolescence. Postgrad Med 50:230, 1971 |
|
LaVecchia C, Morris HB, Draper GJ: Malignant ovarian tumors in childhood in Britain. Br J Cancer 48:363, 1983 |
|
Christopher C: Ovarian neoplasms in childhood and adolescence. Ala J Med Sci 9:318, 1972 |
|
Lindedue BG, du Toit JP, Muller MM, et al: Ultrasonographic criteria for the conservative management of antenatally recognized fetal ovarian cysts. J Reprod Med 33:196, 1988 |
|
Groeber WR: Ovarian tumors during infancy and childhood. Am J Obstet Gynecol 86:1021, 1963 |
|
Towne B, Mahour GH, Wooley MM, et al: Ovarian cysts and tumors in childhood. J Pediatr Surg 10:311, 1975 |
|
Breen JL, Maxson WS: Ovarian tumors in children and adolescents. Clin Obstet Gynecol 20:607, 1977 |
|
Anteby SO, Mor-Josef S, Schenker JG: Ovarian cancer in the young. Eur J Gynaecol Oncol 6:41, 1985 |
|
Brodeuri GM, Howarth CB, Pratt CB, et al: Malignant germ cell tumors in 57 children and adolescents: A review of cases. Cancer 48:1890, 1981 |
|
Ehren IM, Mahour GH, Isaacs H: Benign and malignant ovarian tumors in children and adolescents: A review of cases. Am J Surg 147:339, 1984 |
|
Golladay ES, Mollit DL: Ovarian masses in child and adolescent. South Med J 76:954, 1983 |
|
Harms D, Janig U: Immature teratomas of childhood: Report of 21 cases. Pathol Res Pract 179:388, 1985 |
|
Hernandez E, Rosenberg NB, Parmley TH: Mucinous cystadenoma in a premenarchal girl. South Med J 75:1265, 1982 |
|
Hong SJ, Lurain JR, Tsukada Y, et al: Cystadenocarcinoma of the ovary in a 4 year old: Benign transformation during therapy. Cancer 45:2227, 1980 |
|
Jereb B, Goulouth R, Harlicek S: Ovarian cancer in children and adolescents: A review of 15 cases. Med Pediatr Oncol 3:339, 1977 |
|
Kullendorf CM: Malignant ovarian teratoma in childhood. Z Kinderchir 5:350, 1983 |
|
Lack EE, Perez-Atayde AR, Murthy AS, et al: Granulosa-theca cell tumors in premenarchal girls: A clinical and pathologic study of 10 cases. Cancer 48:1846, 1981 |
|
Lessing JB, Michowitz M, Baratz M: Granulosa-theca cell tumor in a one year old infant. Acta Obstet Gynecol Scand 64:345, 1985 |
|
Morris HB, LaVecchia C, Draper GJ: Malignant epithelial tumors of the ovary in childhood: A clinicopathological study of 13 cases in Great Britain 1962–1978. Gynecol Oncol 19:290, 1984 |
|
Pysher TJ, Hitch DC, Krous HF: Bilateral juvenile granulosa cell tumor in a 4 month old. Am J Obstet Gynecol 143:870, 1982 |
|
Rotmensch J, Woodruff JD: Lymphoma of the ovary: Report of twenty new cases and update of previous series. Am J Obstet Gynecol 143:870, 1982 |
|
Raney RB, Sinclair L, Uri A, et al: Malignant ovarian tumors in children and adolescents. Cancer 59:1214, 1987 |
|
Shawis RM, Elgohary A, Cook RC: Ovarian cysts and tumors in infancy and childhood. Ann R Coll Surg Engl 67:17, 1985 |
|
Weinblatt ME, Ortega JA: Treatment of children with dysgerminoma of the ovary. Cancer 48:2608, 1982 |
|
Yakushiji M, Matsukuma T, Abe M, et al: Ovarian tumors in children and adolescents less than 20 years of age. Acta Obstet Gynaecol Jpn 33:833, 1981 |
|
Zalondek C, Norris HJ: Granulosa tumor of the ovary in children: A clinical and pathologic study of 32 cases. Am J Surg Pathol 6:513, 1982 |
|
Moore JG, Schifrin BS, Erez S: Ovarian tumors in infancy, childhood and adolescence. Am J Obstet Gynecol 99:913, 1967 |
|
Cronen PW, Nagaraj HS: Ovarian tumors in children. South Med J 81:464, 1988 |
|
Khanna S, Arya NC, Gupta IM, et al: Germ cell tumors in Indian children. J Surg Oncol 37:235, 1988 |
|
Haefiner HG, Roberts JA, Schmidt RW: The university experience of clinical and pathological findings of ovarian neoplasms in children and adolescents. Adolesc Pediatr Gynecol 5:182, 1992 |
|
Warner BW, Kuhn JC, Barr U: Conservative management of larger ovarian cysts in children: The value of serial pelvic ultrasonography. Surgery 112:749, 1992 |
|
Skinner MA, Schlatter MG, Heifetz SA, et al: Ovarian neoplasms in children. Arch Surg 128:849, 1993 |
|
Brown MF, Hebra A, McGeehin K, et al: Ovarian masses in children: A review of 91 cases of malignant and benign masses. J Pediatr Surg 28:930, 1993 |
|
Schuck R, Knetsch J, Deeg KH, et al: Diagnosis and treatment of tumors of the adnexes in childhood and adolescence. Monattsschr Kinderheilkd 138:279, 1990 |
|
Breen JL, Neubecker RD: Ovarian malignancy in children with special reference to the germ cell tumors. Ann N Y Acad Sci 142:658, 1967 |
|
Elin S: Malignant ovarian tumors in childhood. J Pediatr Surg 8:539, 1973 |
|
Williams SD, Gershenson DM, Horowitz CJ, et al: Ovarian germ cell and stromal tumors. In Hoskins WJ, et al (ed): Principles and Practice of Gynecologic Oncology. p 987, Philadelphia, JB Lippincott, 1992 |
|
Adelman S, Benson C, Hertzler J: Surgical lesions in infancy and childhood. Surg Gynecol Obstet 141:219, 1975 |
|
Thind CR, Carty HM, Pilling DW: The role of ultrasound in the management of ovarian masses in children. Clin Radiol 40:180, 1989 |
|
Slayton RE: Management of germ cell and stromal tumors of the ovary. Semin Oncol 11:299, 1984 |
|
Williams SD: Current management of ovarian germ cell tumors. Oncology 8:53, 1994 |
|
Williams S, Blessing JA, Liao SY, et al: Adjuvant therapy of ovarian germ cell tumors with cisplatin, etoposide, and bleomycin: a trial of the Gynecologic Oncology Group. J Clin Oncol 12:701, 1994 |
|
Gershenson DM: Chemotherapy of ovarian germ cell tumors and sex cord tumors. Semin Surg Oncol 10:290, 1994 |
|
Breen JL, Neubecker RD: Malignant teratoma of the ovary. Obstet Gynecol 21:669, 1963 |
|
Kurman RJ, Norris HJ: Malignant germ cell tumor of the ovary. Hum Pathol 8:5, 1977 |
|
Slayton RE, Park RC, Silverberg SG, et al: Vincristine, dactomycin, and cyclophosphamide in the treatment of malignant germ cell tumors of the ovary: A Gynecologic Oncology Group study. Cancer 56:243, 1985 |
|
Williams S: Treatment of germ cell tumors of the ovary. Semin Oncol 18:292, 1991 |
|
Williams SD, Blessing J, Slayton R, et al: Ovarian germ cell tumors: Adjuvant trials of the Gynecologic Oncology Group. Proc Am Soc Clin Oncol 8:150, 1989 |
|
Williams S, Birch R, Einhorn LH, et al: Treatment of malignant ovarian germ cell tumors with PVB. Proc Am Soc Clin Oncol 3:175, 1984 |
|
Changiri A, Smith J, Van Eys J: Improved prognosis in children with ovarian cancers following modified VAC chemotherapy. Cancer 45:1234, 1978 |
|
Jacobs AJ, Harris M, Deppe G, et al: Treatment of recurrent and persistent germ cell tumors with cisplatin, vinblastine and bleomycin. Obstet Gynecol 59:129, 1982 |
|
Pinkerton CR, Pritchard J, Spitz L: High complete response rate in children with advanced germ cell tumors using cisplatin-containing chemotherapy. J Clin Oncol 4:194, 1986 |
|
Smales E, Peckham MJ: Chemotherapy of germ cell ovarian tumors. Eur J Cancer Clin Oncol 23:469, 1987 |
|
Gershonson DM, Del Junco G, Herson J, et al: Endodermal sinus tumor of the ovary: The M.D. Anderson Experience Obstet Gynecol 61:194, 1983 |
|
Abell MR, et al: Ovarian neoplasms in childhood and adolescence. Am J Obstet Gynecol 93:850, 1965 |
|
Colombo N, Sessa C, Landoni F, et al: Cisplatin, vinblastine, and bleomycin combination chemotherapy in metastatic granulosa cell tumor of the ovary. Obstet Gynecol 67:265, 1986 |
|
Young RH, Lawrence WD, Scully RE: Juvenile granulosa cell tumor—another neoplasm associated with abnormal chromosomes and ambiguous genitalia. Am J Surg Pathol 9:737, 1985 |
|
Norris H, Taylor H: Prognosis of granulosa–theca cell tumors of the ovary. Cancer 21:255, 1968 |
|
Pedowitz P, O’Brien FB: Arrhenoblastoma of the ovary. Obstet Gynecol 16:62, 1960 |
|
Thigpen T, Blessing J, Ball H, et al: Phase II trial of taxol as a second-line therapy for ovarian carcinoma: A Gynecologic Oncology Group Study. Proc Am Soc Clin Oncol 9:156, 1990 |