Molecular Biology of Gynecologic Cancer
Authors
INTRODUCTION
Proto-oncogenes are deoxyribonucleic acid (DNA) sequences found in the genomes of normal and neoplastic tissues that are quite similar to the transforming genes of ribonucleic acid (RNA) tumor viruses. Proto-oncogenes exhibit striking evolutionary conservation, being found in a large number of phylogenetically diverse organisms ranging from Drosophila flies to humans. More than 40 proto-oncogenes have been described, and this list will undoubtedly continue to expand. Protooncogenes encode growth factors, growth factor receptors, and proteins that regulate cell growth and differentiation. Proto-oncogenes are usually grouped according to the primary function or sequence homology of their protein products (Table 1).
TABLE 1. Proto-Oncogene Classification
| Gene Product or Function | Proto-Oncogene |
Growth factors | FGF growth factor | fgf-5, hst, int-2 |
| PDGF B-chain | sis |
Transmembrane receptors | EGF receptor | erbB |
| EGF-related-receptor | HER-2/neu |
| CSF receptor | fms |
| Stem cell receptor | kit |
| Nerve growth factor receptor | trk |
Inner membrane receptors | Plasma membrane signal transducer | bcl-2 |
| GTP-binding/GTPase activity | Ha-ras, Ki-ras, N-ras |
| Other | fgr, lck, src, yes |
Cytoplasmic messengers | Tyrosine/kinase activity | crk |
| Serine/threonine kinase activity | cot, pim-1, mos, raf/mil |
Nuclear transcription | DNA-binding proteins | erbB1, ets-1, ets-2, fos, gil-1, jun, rel, ski, vav, lyl-1, |
factors |
| maf, myb, myc, L-myc, N-myc, spi-1, evi-1 |
CSF = colony-stimulating factor; EGF = epidermal growth factor; FGF = fibroblast growth factor;
GTP = guanosine triphosphate; GTPase = guanosine triphosphatase;
PDGF = platelet-derived growth factor
Tumor suppressor genes are a second major group of genes that regulate cell growth. These genes, which include the p53 gene and the retinoblastoma gene, encode DNA-binding proteins that normally act to inhibit cellular proliferation.
Proto-oncogenes and tumor suppressor genes encode proteins that play important roles in every step of the intracellular signal transduction system and permit the cell to respond to external stimuli in a specific, coordinated, and controlled fashion (Fig. 1). The net result of the expression of proto-oncogenes and tumor suppressor genes is the regulation of cell growth and differentiation.
|
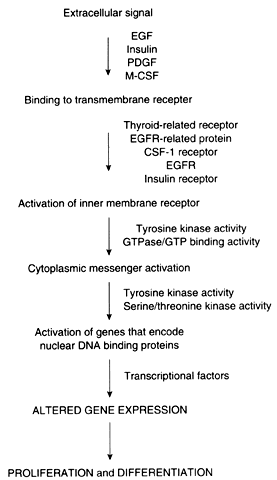
Membrane receptors bind extracellular molecules, such as growth factors and steroid hormones, initiating a cascade of events that culminates in altered gene expression. These receptors are often linked to inner membrane receptors, perhaps the most notable of which are encoded by the ras gene family. The ras genes encode proteins that exhibit guanosine triphosphate (GTP) binding or guanosine triphosphatase (GTPase) activity, or both. These proteins, collectively designated p21, are attached to the inner surface of the cell membrane. These proteins couple membrane receptors to a number of signaling systems, including cyclic adenosine monophosphate and the inositol phospholipid pathway.
Cytoplasmic messenger systems transmit external signals from the cell membrane to the nucleus. This part of the regulatory network involves protein phosphorylation activity and is the target of substances such as platelet-derived growth factor, fibroblast growth factor, epidermal growth factor, colony-stimulating factor, and insulin-like growth factor (see below).
Numerous proto-oncogenes have been identified that encode nuclear DNA-binding proteins that regulate transcription of other genes. Typically, more than one gene acts in concert with several other genes to trigger a change in proliferation. As an example, increased expression of c-jun and c-fos, followed by an increase in c-myc, is required for the transition from the G0 to the G1 phase of the cell cycle. In contradistinction, the p53 tumor suppressor gene encodes a protein that normally acts to inhibit cellular proliferation. Expression of the p53 gene also increases in cells that have sustained DNA damage so that the damage is not passed to daughter cells before repair.
Data derived from in vitro and in vivo models clearly demonstrate that proto-oncogenes, acting as a subset of a larger group of genes, play crucial roles in the regulation of cellular proliferation and differentiation. Studies of the placenta and endometrium are of particular interest to the gynecologist. In the placenta, the trophoblast cell population changes from one dominated by the “pseudo-malignant” cytotrophoblast to one composed predominantly of the highly differentiated syncytiotrophoblast. This transition is accompanied by discrete changes in the expression of several proto-oncogenes. The expression of c-fms, confined to the placenta and extraembryonic membranes, exhibits a progressive increase in expression during prenatal development in the mouse.1 In contradistinction, the expression of c-fos is approximately 15-fold greater in cytotrophoblasts than in syncytiotrophoblasts.2 The transcription of c-myc appears to be directly associated with proliferative activity of the trophoblast.3, 4, 5 Peak c-myc messenger RNA expression occurs at 4 to 6 weeks' estimated gestational age, followed by a rapid decline to barely detectable levels at term. The expression of the Ha-ras proto-oncogene demonstrates little variation with gestational age in placental tissue.
Normal endometrium exhibits cyclic proliferation, differentiation, and exfoliation in response to estrogen and progesterone. These events, which collectively characterize the menstrual cycle, are accompanied by discrete changes in the expression of myc, ras, and fos. Interestingly, the expression of these genes varies not only with the phase of the menstrual cycle, but between the glandular and stromal cell compartments as well.6 Cell- and tissue-specific changes in proto-oncogene expression also have been documented in the developing embryo.7, 8, 9
MALIGNANT TRANSFORMATION
In view of the crucial role that proto-oncogenes and tumor suppressor genes play in normal cell growth and differentiation, it is a logical corollary that the abnormal expression of these genes is causally related to the uncontrolled cellular proliferation that characterizes the neoplastic phenotype. Cellular transformation requires the mutation of several genes via a multistep process. Genetic mutations can result from gene amplification, point mutations, insertional mutagenesis, and deletions or rearrangements, or both (Fig. 2).
{kind=link}
Gene amplification, in which the copy number of the gene is increased, is readily detected by southern hybridization analysis. Amplification can result in enhanced gene expression by increasing the amount of template DNA available for transcription. Proto-oncogene amplification is a relatively common event in gynecologic malignancies. In most cases, enhanced oncogene expression accompanies oncogene amplification.
Point mutations, in which the codon sequence is altered, can result in altered gene action by qualitatively altering the gene product. The ras proto-oncogene and the p53 tumor suppressor gene are two of the best-studied genes in gynecologic malignancies that exhibit point mutations. Point mutations of the ras gene occur in approximately 15% of solid tumors. The replacement of a single amino acid in the ras gene product as the result of a base mutation at codon 12, 13, or 61 results in the synthesis of a p21 ras protein with increased transforming ability.10 The mutated p21 protein exhibits decreased GTPase activity, and the bound GTP is hydrolyzed to guanosine diphosphate more slowly.11 As a consequence, the mutant protein is constitutively activated, and a continuous signal is transduced to the nucleus.
Point mutations of the p53 gene occur at preferential “hot spots” that coincide with the most highly conserved regions of the gene. Loss of normal p53 gene activity removes one of the endogenous “brakes” on cellular proliferation. In addition, point mutations of the p53 gene impair the ability of the cell to repair DNA damage before entry into the S-phase of the cell cycle. As an example, irradiated cells normally exhibit a transient S-phase arrest as p53 gene expression increases and radiation-induced DNA damage is repaired. When a mutated p53 gene is present, the delay at the G1-S checkpoint does not occur, and the DNA damage is passed to the daughter cells. Mutation of the p53 gene is the most frequent genetic alteration found in solid tumors.
Insertional mutagenesis occurs when the regulation of gene expression is altered when DNA sequences that control transcription are deleted, transposed, or placed under the control of a new DNA sequence. Human papillomavirus (HPV), implicated as an important cofactor in the etiology of squamous cell dysplasia and carcinoma, occasionally integrates near the c-myc oncogene during incorporation into the host genome. Juxtaposition of HPV genetic sequences near c-myc has been associated with increased expression of this gene.12 The HeLa cell line contains integrated HPV18 near the c-myc oncogene, which is also overexpressed. In situ hybridization analysis indicates that multiple copies of this virus also integrate near the c-abl and c-sis oncogenes.13
Deletions and rearrangements reflect gross changes in the DNA template that result in the synthesis of a markedly altered protein product.
CERVICAL CANCER
The transition from normal cervical epithelium to dysplasia to invasive disease is well-defined histologically and provides a useful framework upon which molecular alterations can be evaluated. To date, molecular biologic studies have focused primarily on genetic alterations present in invasive cervical cancer.
Structural alterations of the Ha-ras gene reported to occur in cervical cancer include allelic deletions and point mutations. Riou and associates14 reported that Ha-ras exhibited allelic loss in the three variable tandem repeats in 36% of heterozygous tumors. Point mutations of Ha-ras, codon 12, were found in 8 of 76 cervical carcinoma specimens, the majority of which were from patients with advanced-stage disease. In addition, 40% of the tumors with codon-12 mutations also exhibited allelic deletions. Other investigators, however, have failed to demonstrate point mutations in a significant proportion of cervical neoplasms.15, 16
The c-erbB gene, which encodes the epidermal growth factor receptor (EGF) receptor, can be overexpressed in cervical neoplasms. Elevated amounts of EGF receptor have been found in severe dysplasia and invasive cervical carcinoma specimens.17 Kohler and colleagues18 performed a comprehensive analysis of EGF receptor in cervical carcinoma specimens using a radioligandbinding assay, northern hybridization, and immunohistochemistry. Thirty-four of 42 specimens expressed EGF receptor in high amounts (greater than 10 fmol/mg specific binding) in 42%. Burchuck and co-workers19 reported that squamous cell carcinomas of the cervix consistently expressed moderate to heavy immunohistochemical staining for EGF receptor.
The c-myc proto-oncogene also has been studied in cervical cancer.20, 21,22, 23 Amplification has been reported to occur in 30% to 80% of cases. Of particular interest are the reports by Riou and associates,24, 25, 26, 27, 28 who correlated amplification and expression of the c-myc gene with prognosis in early-stage cervical carcinoma. Using northern hybridization and slot blot analysis, they found c-myc overexpression in 33% of 93 cervical carcinoma specimens. They found no apparent relationship between c-myc expression and histology, tumor differentiation, status of the surgical margins, presence or absence of lymphovascular space involvement, or endocervical extension. They considered overexpression of c-myc in lymph node—negative patients, however, to be predictive of subsequent relapse. Three-year survival for patients with normal c-myc expression and negative pelvic lymph nodes was 93%, compared with only 51% for patients with overexpression of the c-myc oncogene and negative lymph nodes. Patients with nodal metastases at the time of radical hysterectomy who had tumors with normal c-myc expression had a 3-year survival of 44%, compared with only 15% for patients with tumors exhibiting overexpression of c-myc.
The expression of c-myc in paraffin-embedded tumor biopsies from patients with advanced-stage disease does not appear to be a prognostic indicator. Symonds and associates29 found that immunohistochemical positivity for c-myc was not associated with time to local recurrence or development of metastases or survival in 55 patients with stage III or stage IV cervix cancer who were treated with neoadjuvant chemotherapy followed by radiation therapy.
The p53 tumor suppressor gene is not commonly mutated in cervical neoplasms. Busby-Earle and colleagues30 studied 57 cases of cervical cancer and found p53 mutations in 2 cases. Helland and co-workers31 evaluated 92 cervix neoplasms by PCR and DGGE and found only two examples of p53 mutations. When mutations are present, they are often found in HPV-negative neoplasms. Fujita and associates32 found p53 mutations in 2 of 36 cervical neoplasms.
Immunohistochemical studies of p53 have revealed detectable expression in a variable proportion of cases. Busby-Earle and colleagues reported p53 positivity in 8 of 57 cervical neoplasms, and the pattern of staining was described as sparse.30 Bosari and co-workers, however, reported p53 positivity in 74% of biopsy specimens from invasive squamous cell carcinomas of the cervix.33 These observations may initially seem inconsistent with the reported rarity of p53 gene mutations in cervical cancer. It is generally presumed that the very short half-life of the normal p53 protein and the small amounts present in normal cells precludes detection by immunohistochemical methods; however, immunohistochemical detection of normal p53 protein has been reported, in colon and lung carcinoma cell lines in the absence of mutations.34, 35 Interruption of the degradation of normal p53 protein is the suggested mechanism.36
With respect to dysplastic lesions, Pinion and associates evaluated the amount of Ha-ras p21 protein in cervical biopsy specimens using fluorescent immunohistochemistry with quantitation by automated image analysis.17 Elevated amounts of the Ha-ras protein product were found in severe dysplasia and invasive carcinoma biopsy specimens as compared with normal cervical tissues. Sagae and colleagues37 also reported that ras p21 protein expression increased with lesion severity.
Berchuck and co-workers reported that the basal keratinocytes of normal cervical epithelium express EGF receptor.19 They found that keratinocyte differentiation was associated with a decrease in staining intensity of the EGF receptor protein. As one might anticipate, carcinoma in situ lesions, characterized by full-thickness loss of differentiation, exhibited full-thickness expression of EGF receptor.
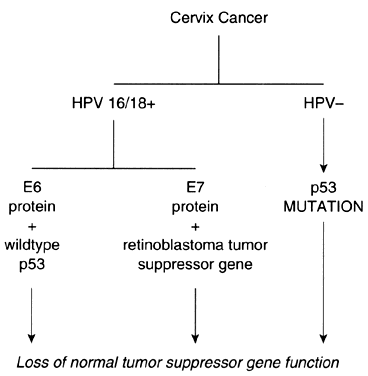
Investigations that have focused on the oncogenic potential of HPV in cervical neoplasms have provided a framework upon which some of the observations concerning oncogenes and tumor suppressor genes can be unified (Fig. 3). Although there are no data to indicate that HPV by itself is capable of causing either cervical dysplasia or cancer, there is abundant evidence to suggest that it is an important cofactor in the pathogenesis of cervical neoplasms. When found in invasive squamous cell carcinomas of the cervix, HPV16 and HPV18 are integrated into the host genome. On occasion, the HPV may insert near chromosomal fragile sites. Some of these sites are also in close proximity to cellular oncogenes, supporting the hypothesis that insertional mutagenesis may be one mechanism of HPV-related oncogenesis.
{kind=link}
Couturier and associates38 demonstrated that HPV sequences were localized to chromosome band 8q24.1, which is also the location of c-myc. In each instance, the c-myc gene was structurally altered or overexpressed. Hori and colleagues39 also reported that HPV16 was integrated in a fragile site near 8q24.1 in a cell line established from a small cell carcinoma of the cervix. Popescu and DiPaolo40 have also reported that fragile sites are preferential locations for HPV integration. Apart from insertional mutagenesis, specific HPV protein products appear to exert inhibitory effects on the p53 and retinoblastoma tumor suppressor gene proteins. HPV16 and HPV18 encode proteins that can bind to the gene products of tumor suppressor genes. As a consequence, the normal activity of these tumor suppressor gene products, which is the inhibition of cellular proliferation, is negated. Interaction of HPV16 E7 protein with the retinoblastoma gene product results in inactivation of its function.41 The E6 protein of HPV16 and HPV18 interacts with the p53 gene product, accelerating its degradation.42, 43 The E6-p53 protein interaction is an appealing explanation for the role of HPV in the etiology of cervical dysplasia and neoplasia, although its biologic significance must be clarified.
Although HPV is present in the majority of squamous cell carcinomas of the cervix, there are well-documented, albeit infrequent, instances of HPV-negative cervical neoplasms.44, 45 In some of these cases, the p53 protein contains a point mutation that results in a gene product with a prolonged half-life. As a consequence of these mutations, the p53 protein interaction with cellular heat shock proteins is enhanced, and confirmation changes occur that abrogate the p53 genes' product activity as an inhibitor of cellular proliferation. Not all HPV-negative cervical neoplasms, however, contain a mutated p53 gene. In addition, there is no apparent correlation between p53 levels and tumorigenicity. The observation that reintroduction of an intact single copy of chromosome 11 into HeLa cells or SiHa cells reverses their tumorigenicity suggests the presence of an important, but unidentified, tumor suppressor gene that may play a crucial role in the development of cervical cancer.46, 47
ENDOMETRIAL ADENOCARCINOMA
Like cervical cancer, endometrial cancer is preceded by precursor lesions that have been histologically characterized. Recent investigations have focused on the molecular genetic changes in both adenocarcinoma and hyperplasia in an effort to determine the genetic mutations that may be causally related to the etiology of this disease.
Point mutations of the ras gene family commonly occur in endometrial adenocarcinoma and atypical adenomatous hyperplasia. The published data are summarized in Table 2.48, 49, 50, 51, 52, 53, 54 It appears that ras mutations in endometrial adenocarcinoma most commonly involve the second nucleotide of codon 12 of the Ki-ras gene. Mutations of the Ki-ras gene may signify an “early” event in endometrial carcinogenesis because it is also present in precursor lesions. At this time, abnormal expression and mutations of the ras gene family do not appear to be of prognostic significance. Scambia and co-workers,55 however, reported that estrogen receptor—positive neoplasms express higher levels of p21 than estrogen receptor—negative tumors. They observed a similar trend between p21 and the progesterone receptor. Interestingly, they found p21 expression to be unrelated to EGF receptor levels.
TABLE 2. Ki-ras Codon-12 Mutations in Endometrial Neoplasms
| Hyperplasia |
| ||
| Simple | Complex | Atypical | Adenocarcinoma |
Susaki et al (1993) |
|
|
| 15/84 |
Enomoto et al (1991) | 0/6 | 0/12 | 2/16 | 4/10 |
Enomoto et al (1990) |
|
|
| 7/19 |
Mizuuchi et al (1992) |
|
|
| 5/49 |
Fujimoto et al (1993) |
|
|
| 10/45 |
Lester and Cauchi (1990) |
|
|
| 2/12 |
Ignar-Trowbridge et al (1992) |
|
|
| 3/30 |
Published data concerning p53 mutations in endometrial adenocarcinoma are summarized in Table 3.56, 57, 58, 59, 60, 61 Mutations of the p53 tumor suppressor gene have not been described in precursor lesions such as atypical adenomatous hyperplasia, suggesting that this mutation is unlikely to be an early event in the development of endometrial adenocarcinoma.
TABLE 3. p53 Mutations in Endometrial Carcinoma
Honda et al (1993) | 4/42 |
Kohler et al (1993) | 3/30 |
Kohler et al (1992) | 22/107 |
Bur et al (1992) | 14/47 |
Naito et al (1992) | 2/7 |
Okamoto et al (1991) | 3/24 |
Amplification of the c-myc oncogene appears to be a relatively common event in endometrial adenocarcinoma, occurring in approximately 30% of cases.62 There are no data concerning the frequency of this mutation in precursor lesions.
The c-erbB and HER-2/neu genes commonly exhibit abnormal expression in endometrial adenocarcinoma. In one study of 34 endometrial adenocarcinoma specimens,63 21 exhibited normal amounts of EGF receptor. In 13 specimens, the amount of EGF receptor was decreased, the greatest reduction occurring in poorly differentiated lesions. In contradistinction, another immunohistochemical study of 69 cases of endometrial carcinoma64 demonstrated EGF receptor expression in 49% of the specimens. EGF receptor positivity was correlated with nonendometrioid histologies, metastatic disease at the time of diagnosis, and decreased survival. HER-2/neu overexpression was present in 59% of specimens and was significantly associated with depth of myometrial invasion.
Other studies have also demonstrated a role for HER-2/neu expression as a prognostic indicator. Berchuck and associates65 studied 95 endometrial carcinomas using immunohistochemistry and found that overexpression of HER-2/neu correlated with advanced-stage disease and decreased survival. Hetzel and colleagues66 reported similar results after evaluating HER-2/neu immunohistochemical expression in 247 endometrial carcinoma specimens. Strong overexpression of HER-2/neu was correlated with decreased progression-free survival and decreased overall survival.
Interestingly, overexpression of the HER-2/neu gene product has been associated with the absence of the estrogen receptor.65 This may represent one way in which the neoplasm circumvents its requirement for estrogen-dependent growth.
The c-fms proto-oncogene encodes the colony-stimulating factor (CSF) receptor. Baiocchi and co-workers67 reported that five of nine tumor samples expressed M-CSF and that six of nine samples expressed the c-fms transcript. Leiserowitz and associates68 demonstrated that the amount of c-fms transcript increases from secretory to proliferative to hyperplastic endometrium. The highest levels were found in adenocarcinoma specimens. It has been proposed that an interaction between M-CSF (the ligand) and c-fms (which encodes the ligand receptor) might contribute to tumor progression and metastatic disease via an autocrine mechanism, in which the tumor stimulates itself to proliferate. In vitro studies have supported this hypothesis.69 CSF receptor—positive cell lines were found to exhibit increased invasiveness in the amniotic basement membrane model compared with CSF receptor—negative cell lines after stimulation with CSF.69 Overexpression of c-fms has been correlated positively with adverse clinical prognostic indicators.67, 68, 70
EPITHELIAL OVARIAN CANCER
Unlike cervical cancer and endometrial cancer, epithelial ovarian cancer does not have an accepted identifiable precursor lesion. In addition, most studies of molecular genetic alterations in ovarian cancer have been performed with tissue from advanced-stage neoplasms. For these reasons, it has been more difficult to differentiate between molecular genetic alterations that are causally related to disease development and alterations that may simply reflect the inherent genetic instability of advanced neoplasms. These difficulties notwithstanding, a number of genetic alterations have been identified in epithelial ovarian cancer.
Amplification of the c-myc gene has been described in approximately one third of advanced-stage epithelial ovarian neoplasms.71, 72 Sasano and colleagues73 studied the immunocytochemical localization of c-myc in 18 archival ovarian carcinoma specimens; 14 serous carcinomas were positive for c-myc expression, and 4 of 4 mucinous carcinomas exhibited c-myc expression.
Elevated levels of ras p21 protein have been reported by several investigators.74, 75, 76, 77 Amplification of the ras gene has also been reported.78, 79, 80, 81 Prognostic roles for ras gene amplification, point mutation, or overexpression have not been identified. In addition, the role of ras gene abnormalities in the initiation and progression of epithelial ovarian cancer remains to be determined.
The HER-2/neu gene is commonly amplified in ovarian cancer. Slamon and co-workers82 reported that this gene was amplified in 31 of 120 cases of epithelial ovarian cancer. Following this study, a number of reports have been published concerning the expression of HER-2/neu in epithelial ovarian cancer (Table 4).83, 84, 85, 86, 87, 88, 89, 90 It appears that HER-2/neu positivity is more common in well-differentiated lesions, endometrioid cell types, and advanced-stage neoplasms.85, 89 The prognostic significance of HER-2/neu overexpression it still being debated; however, the majority of reports suggest little, if any, role.
TABLE 4. HER-2/neu as a Prognostic Indicator in Epithelial Ovarian Neoplasms
Authors | Prognostic Indicator |
Slamon et al (1989) | Yes |
Berchuck et al (1990) | Yes |
Haldane et al (1990) | No |
Seidman et al (1992) | No |
Kacinski et al (1992) | No |
Garuti and Genazzani (1991) | No |
Rubin et al (1993) | No |
Huettner et al (1992) | No |
The c-erbB proto-oncogene, which encodes EGF receptor, is commonly overexpressed in ovarian carcinoma. Normally, EGF receptor mediates cellular proliferation by binding EGF or transforming growth factor-α (TGF-α).
The c-fms proto-oncogene encodes the receptor for CSF. Baiocchi and associates67 found elevated levels of mRNA c-fms transcripts in 78% of 17 stage III ovarian cancers. With the exception of one neoplasm, all of these lesions also expressed M-CSF, which is the ligand for the CSF receptor. Kacinski and colleagues90 found that 14 of 14 ovarian cancer specimens expressed fms transcripts and that 7 also expressed M-CSF. It has been suggested that c-fms and M-CSF contribute to autocrine and paracrine induction of neoplastic proliferation and progression in ovarian cancer. Abnormal expression of c-fms has not been demonstrated to be of prognostic significance in multivariate analyses.
Point mutations of the p53 gene have been studied in some detail in ovarian cancer. Kihana and co-workers91 studied 14 epithelial ovarian cancers and found abnormalities of the p53 gene in 10. Four of these mutations were in exon 7, two were in exon 5, two were in exon 2, one was in exon 8, and one was in exon 6. The point mutations in the p53 gene are typically transitions (purine to purine or pyrimidine to pyrimidine) and suggest mutations resulting from spontaneous errors in DNA synthesis and repair, rather than carcinogen-induced transversions (purine to pyrimidine and vice versa).
Mazars and associates92 studied 30 primary ovarian cancer specimens and found that 11 had a mutated allele. Point mutations were restricted to exons 5 and 7. Both the primary lesion and the metastatic lesions from the same patient exhibited the same codon changes, indicating the clonality of epithelial ovarian cancer. Similar results indicating the clonality of epithelial ovarian cancer have been published by other investigators.93, 94
In general, point mutations of the p53 gene result in a protein product with a prolonged half-life, allowing p53 protein to be detected by immunohistochemical methods. Marks and colleagues95 examined p53 expression in 107 epithelial ovarian neoplasms, of which 54 exhibited overexpression of p53. There was no correlation between overexpression and stage, grade, or the ability to resect the tumor optimally.
Kohler and co-workers analyzed 53 stage I and II epithelial ovarian neoplasms and reported overexpression in 15.96 Stage IC and II neoplasms, poorly differentiated lesions, or neoplasms larger than 10 cm were more likely to overexpress p53 compared with neoplasms without these features, although the differences were not statistically significant. They found that overexpression of p53 was not associated with recurrence or survival. Berchuck and associates97 found overexpression of the p53 gene in only 2 of 49 ovarian neoplasms of low malignant potential.
The etiologic role of p53 in ovarian cancer has not been determined, although there is no evidence based on immunohistochemical analyses to suggest that it is an early event. Although p53 is often overexpressed, particularly in advanced-stage and poorly differentiated neoplasms, the true incidence of p53 mutations is not known. Kupryanczyk and colleagues98 have clearly demonstrated that immunohistochemical evidence of p53 expression in epithelial ovarian cancer provides an underestimate of p53 mutations compared with the results obtained by molecular genetic analysis of exons 2 to 11. Nonsense mutations, splicing mutations, and deletions might not result in p53 accumulation.
A number of genetic mutations have been described in ovarian cancer, but the specific alterations that are causally involved in the etiology of this disease have not been identified. Presumably, repeated ovulation with disruption of the germinal epithelium, followed by activation of cellular repair mechanisms, provides ample opportunity for somatic gene mutations and deletions to occur.99 Recently described in vitro models will promote testing of this hypothesis.100
REFERENCES
Muller R, Slamon DJ, Adamson ED et al: Transcription of c-onc genes c-ras and c-fms during mouse development. Mol Cell Biol 3: 1062, 1983 |
|
Muller R, Verma IM, Adamson ED: Expression of c-onc genes: c-fos transcripts accumulate to high levels during development of mouse placenta, yolk sac, and amnion. EMBO J 2: 679, 1983 |
|
Pfeiffer-Ohlsen S, Goustin AS, Rudnert J et al: Spatial and temporal pattern of cellular myc oncogene expression in developing human placenta: Implications for embryonic cell proliferation. Cell 38: 585, 1984 |
|
Goustin AS, Betsholtz C, Pfeiffer-Ohlson S et al: Co-expression of sis and myc proto-oncogene in developing human placenta suggests autocrine control of trophoblast growth. Cell 41: 301, 1985 |
|
Maruo T, Mochizuki M: Immunohistochemical localization of epidermal growth factor and myc oncogene product in human placenta: Implications for trophoblast proliferation and differentiation. Am J Obstet Gynecol 156: 721, 1987 |
|
Stancel GM, Baker VV, Hyder SM et al: Oncogenes and uterine function. Oxford Rev Biol 15: 1, 1993 |
|
Muller R, Slamon DJ, Tremblay JM et al: Differential expression of cellular oncogenes during prenatal and postnatal development of the mouse. Nature 299: 640, 1982 |
|
Slamon DJ, deKernion JB, Verma IM et al: Expression of cellular oncogenes during embryonic and fetal development of the mouse. Proc Natl Acad Science USA 81: 7141, 1984 |
|
Pfeifer-Ohlson S, Rydnert J, Goustin AS et al: Cell type specific patterns of c-myc expression in developing human embryos. Proc Natl Acad Science USA 82: 5050, 1986 |
|
Sistonen L, Alitalo K: Activation of c-ras oncogenes by mutations and amplification. Ann Clin Res 18: 297, 1986 |
|
Gibbs JB, Sigal IS, Poe M et al: Intrinsic GTPase activity distinguishes normal and oncogenic ras p21 proteins. Proc Natl Acad Science USA 81: 5704, 1984 |
|
Durst M, Croce M, Gissmann L et al: Papillomavirus sequences integrate near cellular oncogenes in some cervical cancers. Proc Natl Acad Sci USA 84: 1070, 1987 |
|
Popescu NC, DiPaolo JA, Amsbaugh SC: Integration sites of human papillomavirus 18 DNA sequences on HeLa cell chromosomes. Cytogenet Cell Genet 44: 58, 1987 |
|
Riou G, Barrois M, Sheng ZM et al: Somatic deletions and mutations of c-Ha-ras gene in human cervical cancers. Oncogene 3: 329, 1988 |
|
Willis G, Jennings B, Ball RY et al: Analysis of ras point mutations and human papillomavirus 16 and 18 in cervical carcinomata and their metastases. Gynecol Oncol 49: 359, 1993 |
|
Koulos JP, Wright TC, Mitchell MF et al: Relationships between c-Ki-ras mutations, HPV type, and prognostic indicators in invasive endocervical adenocarcinomas. Gynecol Oncol 48: 364, 1993 |
|
Pinion SB, Kennedy JH, Miller RW, MacLean AB: Oncogene expression in cervical intraepithelial neoplasia and invasive cancer of the cervix. Lancet 337: 819, 1991 |
|
Kohler M, Janz I, Wintzer HO et al: The expression of EGF receptors, EGF-like factors and c-myc in ovarian and cervical carcinomas and their potential clinical significance. Anitcancer Res 9: 1537, 1989 |
|
Berchuck A, Rodriquez G, Kamel A et al: Expression of epidermal growth factor receptor and HER-2/neu in normal and neoplastic cervix, vulva, and vagina. Obstet Gynecol 76: 381, 1990 |
|
Choo KB, Chong KY, Chou HF et al: Analysis of the structure and expression of the c-myc oncogene in cervical tumor and in cervical tumor-derived cell lines. Biochem Biophys Res Commun 158: 334, 1989 |
|
Baker VV, Hatch KD, Shingleton HM: Amplification of the c-myc proto-oncogene in cervical carcinoma. J Surg Oncol 39: 225, 1988 |
|
Munzel P, Marx D, Kochel H et al: Genomic alterations of the c-myc proto-oncogene in relation to the over-expression of c-erbB2 and Ki67 in human breast and cervix carcinomas. J Cancer Res Clin Oncol 117: 603, 1991 |
|
Ocadiz R, Sauceda R, Cruz M et al: High correlation between molecular alterations of the c-myc oncogene and carcinoma of the uterine cervix. Cancer Res 47: 4173, 1987 |
|
Riou G, Barrois M, Le MG et al: C-MYC proto-oncogene expression and prognosis in early carcinoma of the uterine cervix. Lancet 1: 761, 1987 |
|
Riou G: Proto-oncogene and prognosis in early carcinoma of the uterine cervix. Cancer Surv 7: 441, 1988 |
|
Bourhis J, Le MG, Barrois M et al: Prognostic value of c-myc proto-oncogene over-expression in early invasive carcinoma of the cervix. J Clin Oncol 8: 1789, 1990 |
|
Riou G, Bourhis J, Le MG: The c-myc proto-oncogene in invasive carcinomas of the uterine cervix: Clinical relevance of over-expression in early stages of the cancer. Anticancer Res 10: 1225, 1990 |
|
Riou G, Sherg ZM, Zhou D et al: C-myc and c-Ha-ras proto-oncogenes in cervical cancer: Prognostic value. Bull Cancer 77: 341, 1990 |
|
Symonds RP, Habeshaw T, Paul J et al: No correlation between ras, c-myc, and c-jun proto-oncogene expression and prognosis in advanced carcinoma of the cervix. Eur J Cancer 28A: 1615, 1992 |
|
Busby-Earle RMC, Steel M, Williams ARW et al: p53 mutations in cervical carcinogenesis—Low frequency and lack of correlation with human papillomavirus status. Br J Cancer 69: 732, 1994 |
|
Helland A, Holm R, Kristensen G et al: Genetic alterations of the TP53 gene, p53 protein expression, and HPV infection in primary cervical carcinomas. J Pathol 171: 105, 1993 |
|
Fujita M, Inoue M, Tanizawa O et al: Alterations of the p53 gene in human primary cervical carcinoma with and without human papillomavirus infection. Cancer Res 52: 5323, 1992 |
|
Bosari S, Roncali M, Viale G et al: p53 immunoreactivity in inflammatory and neoplastic diseases of the uterine cervix. J Pathol 169: 425, 1993 |
|
Rodriquez NR, Rowan A, Smith NEF et al: p53 mutations in colorectal cancer. Proc Natl Acad Sci USA 87: 7555, 1991 |
|
Lehman T, Bennett WP, Metcalf RA et al: p53 mutations, ras mutations and p53-heat shock protein complexes in human lung carcinoma cell lines. Cancer Res 51: 4090, 1991 |
|
Wynford-Thomas D: p53 in tumour pathology: Can we trust immunohistochemistry? J Pathol 166: 329, 1992 |
|
Sagae S, Kudo R, Kuzumaki N, et al: Ras oncogene expression and progression in intraepithelial neoplasia of the uterine cervix. Cancer 66: 295, 1990 |
|
Couturier J, Sastre-Garau X, Schneider-Maunoury S et al: Integration of papillomavirus DNA near myc genes in genital carcinomas and its consequences for proto-oncogene expression. J Virol 65: 5434, 1991 |
|
Hori T, Ichimura H, Minamihisamatsu M et al: Chromosomal insertion and amplification of human papillomavirus 16 DNA sequences in a cell line of argyrophil small cell carcinoma of the uterine cervix. Jpn J Cancer Res 82: 371, 1991 |
|
Popescu NC, DiPaolo JA: Preferential sites for viral integration on mammalian genome. Cancer Genet Cytogenet 42: 157, 1989 |
|
Dyson N, Howley PM, Munger K, Harlow E: The human papillomavirus 16-E7 oncoprotein is able to bind to the retinoblastoma gene product. Science 243: 934, 1989 |
|
Werness BA, Levine AJ, Howley PM: Association of human papillomavirus types 16 and 18 E6 proteins with p53. Science 248: 76, 1990 |
|
Scheffner M, Werness BA, Hurbergtse JM et al: The E6 oncoprotein encoded by human papillomavirus types 16 and 18 promotes the degradation of p53. Cell 63: 1129, 1990 |
|
Scheffner M, Munger K, Byrne JC, Howley PM: The state of p53 and retinoblastoma genes in human cervical carcinoma cell lines. Proc Natl Acad Sci USA 88: 5523, 1991 |
|
Crook T, Wrede D, Vousden KH: p53 point mutations in HPV negative human cervical carcinoma cell lines. Oncogene 6: 873, 1991 |
|
Saxon PJ, Srivatson ES, Stanbridge EJ: Introduction of human chromosome 11 via microcell transfer controls tumorigenic expression of HeLa cells. EMBO J 5: 3461, 1985 |
|
Koi M, Morita H, Yamada H et al: Normal human chromosome 11 suppresses tumorigenicity of human cervical tumor cell line SiHa. Mol Carcinog 2: 12, 1989 |
|
Susaki H, Nishii H, Takahashi H et al: Mutation of the Ki-ras proto-oncogene in human endometrial hyperplasia and carcinoma. Cancer Res 53: 1906, 1993 |
|
Enomoto T, Inoue M, Perantoni AO et al: K-ras activation in premalignant and malignant epithelial lesions of the human uterus. Cancer Res 51: 5308, 1991 |
|
Enomoto T, Inoue M, Perantoni AO et al: K-ras activation in neoplasms of the human female genital tract. Cancer Res 50: 6139, 1990 |
|
Mizuuchi H, Nasim S, Kudo R et al: Clinical implications of K-ras mutations in malignant epithelial tumors of the endometrium. Cancer Res 52: 2777, 1992 |
|
Fujimoto I, Shimizu Y, Hirai Y et al: Studies on ras oncogene activation in endometrial carcinoma. Gynecol Oncol 48: 196, 1993 |
|
Lester DR, Cauchi MN: Point mutations at codon 12 of c-K-ras in human endometrial carcinoma. Cancer Lett 51: 7, 1990 |
|
Ignar-Trowbridge D, Risinger JI, Dent GA et al: Mutations of the Ki-ras oncogene in endometrial carcinoma. Am J Obstet Gynecol 167: 227, 1992 |
|
Scambia G, Catozzi L, Benedetti-Panici P et al: Expression of ras p21 oncoprotein in normal and neoplastic human endometrium. Gynecol Oncol 50: 339, 1993 |
|
Honda T, Kato H, Imamura T et al: Involvement of p53 gene mutations in human endometrial carcinoma. Int J Cancer 53: 963, 1993 |
|
Kohler MF, Nichii H, Humphrey PA et al: Mutation of the p53 tumor-suppressor gene is not a feature of endometrial hyperplasia. Am J Obstet Gynecol 169: 690, 1993 |
|
Kohler MF, Berchuck A, Davidoff AM et al: Over-expression and mutations of p53 in endometrial carcinoma. Cancer Res 52: 1622, 1992 |
|
Bur ME, Perlman C, Edelman L et al: p53 expression in neoplasms of the uterine corpus. Am J Clin Pathol 98: 81, 1992 |
|
Naito M, Satake M, Sakai E et al: Detection of p53 gene in human ovarian and endometrial cancers by polymerase chain reaction single strand conformation polymorphism analysis. Jpn J Cancer Res 83: 1030, 1992 |
|
Okamoto A, Sameshima Y, Yamada Y et al: Allelic loss on chromosome 17p and p53 mutations in human endometrial carcinoma of the uterus. Cancer Res 51: 5632, 1991 |
|
Borst MP, Baker VV, Dixon D et al: Oncogene alterations in endometrial carcinoma. Gynecol Oncol 38: 364, 1990 |
|
Reynolds RK, Talavera F, Roberts JA et al: Characterization of epidermal growth factor receptor in normal and neoplastic human endometrium. Cancer 66: 1967, 1990 |
|
Khalifa MA, Mannel RS, Haraway SD et al: Expression of EGFR, HER-2/neu, p53 and PCNA in endometrioid, serous papillary, and clear cell endometrial adenocarcinomas. Gynecol Oncol 53: 84, 1994 |
|
Berchuck A, Rodriquez G, Kinney RB et al: Over-expression of HER-2/neu is associated with advanced stage disease. Am J Obstet Gynecol 164: 15, 1991 |
|
Hetzel DJ, Wilson TO, Keeney GL et al: HER-2/neu expression: A major prognostic factor in endometrial cancer. Gynecol Oncol 47: 179, 1992 |
|
Baiocchi G, Kavanagh JJ, Talpaz M et al: Expression of the macrophage colony stimulating factor and its receptor in gynecologic malignancies. Cancer 67: 990, 1991 |
|
Leiserowitz GS, Harris SA, Subramaniam M et al: The proto-oncogene c-fms is over-expressed in endometrial cancer. Gynecol Oncol 49: 190, 1993 |
|
Filderman AE, Bruckner A, Kacinski B et al: Macrophage colony stimulating factor (CSF-1) enhances invasiveness in CSF-1 receptor positive carcinoma cell lines. Cancer Res 52: 3661, 1992 |
|
Kacinski BM, Carter D, Mittal K et al: High level of expression of c-fms proto-oncogene mRNA is observed in clinically aggressive human endometrial adenocarcinomas. Int J Radiat Oncol Biol Phys 15: 823, 1988 |
|
Baker VV, Borst MP, Dixon D et al: c-myc Amplification in ovarian cancer. Gynecol Oncol 38: 340, 1990 |
|
Sasano H, Garrett CT: Oncogenes in gynecologic tumors. Curr Top Pathol 85: 357, 1992 |
|
Sasano H, Nagura H, Silverburg SG: Immunolocalization of c-myc oncoprotein in mucinous and serous adenocarcinomas of the ovary. Hum Pathol 23: 491, 1992 |
|
Scambia G, Catozzi L, Panici PB et al: Expression of ras oncoprotein p21 protein in normal and neoplastic ovarian tissues: Correlation with histopathologic features and receptors for estrogen, progesterone, and epidermal growth factor receptor. Am J Obstet Gynecol 168: 71, 1993 |
|
Yaginuma Y, Yamashita K, Kuzumaki N et al: ras Oncogene product p21 expression and prognosis of human ovarian tumors. Gynecol Oncol 46: 45, 1992 |
|
Kotylo PK, Michael H, Fineberg N et al: Flow cytometric analysis of DNA content and ras p21 oncoprotein expression in ovarian neoplasms. Int J Gynecol Pathol 11: 30, 1992 |
|
Hiwasa T, Hirono M, Suzuki M et al: Expression and localization of epidermal growth factor receptors and ras oncogene products in gynecologic tumors. Eur J Gynaecol Oncol 13: 241, 1992 |
|
Gukumoto M, Estensen RD, Sha L et al: Association of Ki-ras with amplified DNA sequences, detected in human ovarian carcinomas by a modified in-gel renaturation assay. Cancer Res 49: 1693, 1989 |
|
Masuda H: Tumor specificity of proto-oncogene amplification in human malignant disease. Proceedings of the Annual Meeting of the Japanese Cancer Associates, Vol 46, p 119. Japanese Cancer Associates, 1990 |
|
Vaan't Veer LJ, Hermens R, Van den Berg-Bakker LAM et al: RAS oncogene amplification in human ovarian carcinoma. Oncogene 2: 157, 1988 |
|
Boltz EM, Keford RF, Leary JA et al: Amplification of c-ras Ki oncogene in human ovarian tumors. Int J Cancer 43: 428, 1989 |
|
Slamon DJ, Godolphin W, Jones LA et al: Studies of the HER-2/neu proto-oncogene in human breast and ovarian cancer. Science 244: 707, 1989 |
|
Berchuck A, Kamel A, Whitaker R et al: Over-expression of HER-2/neu is associated with poor survival in advanced ovarian cancer. Cancer Res 50: 4087, 1990 |
|
Haldane JS, Hird V, Hughes CM, Gullick WJ: c-erbB-2 Oncogene expression in ovarian cancer. J Pathol 162: 231, 1990 |
|
Seidman JD, Frisman DM, Norris HJ: Expression of the HER-2/neu proto-s in serous ovarian neoplasms. Cancer 70: 2875, 1992 |
|
Kacinski BM, Mayer AG, King BL et al: NEU protein over-expression in benign, borderline, and malignant ovarian neoplasms. Gynecol Oncol 44: 245, 1992 |
|
Garuti G, Genazzani AR: Human neu oncogene is expressed in endometrial but not in ovarian adenocarcinomas. Cancer 67: 1713, 1991 |
|
Rubin SC, Finstad CL, Wong GY et al: Prognostic significance of HER-2/neu expression in advanced epithelial ovarian cancer: A multivariate approach. Am J Obstet Gynecol 168: 162, 1993 |
|
Huettner PC, Carney WP, Naber SP et al: Neu oncogene expression in ovarian tumors: A quantitative study. Mod Pathol 5: 250, 1992 |
|
Kacinski BM, Carter D, Mittal K et al: Ovarian adenocarcinomas express fms-complementary transcripts and fms antigen, often with coexpression of CSF-1. Am J Pathol 137: 135, 1990 |
|
Kihana T, Tsuda H, Teshima S et al: High incidence of p53 gene mutations in human ovarian cancer and its association with nuclear accumulation of p53 protein and tumor DNA aneuploidy. Jpn J Cancer Res 83: 978, 1992 |
|
Mazars R, Pujol P, Maudelonde T et al: p53 mutations in ovarian cancer: A late event? Oncogene 6: 1685, 1991 |
|
Okamoto A, Sameshima Y, Yakoyama S et al: Frequent allelic losses and mutation of the p53 gene in human ovarian cancer. Cancer Res 51: 5171, 1991 |
|
Jacobs IJ, Kohler MF, Wiseman RW et al: Clonal origin of epithelial ovarian carcinoma: Analysis by loss of heterozygosity, p53 mutation, and X-chromosome inactivation. J Natl Cancer Inst 84: 1793, 1992 |
|
Marks JR, Davidoff AM, Kerns BJ et al: Overexpression and mutation of p53 in epithelial ovarian cancer. Cancer Res 51: 2979, 1991 |
|
Kohler MF, Kerns B-J M, Humphrey PA et al: Mutations and overexpression of p53 in early stage epithelial ovarian cancer. Obstet Gynecol 81: 643, 1993 |
|
Berchuck A, Kohler MF, Hopkins MP et al: Overexpression of p53 is not a feature of benign and early stage borderline epithelial ovarian tumors. Gynecol Oncol 52: 232, 1994 |
|
Kupryanczyk J, Thor AD, Beauchamp R et al: p53 gene mutations and protein accumulation in human ovarian cancer. Proc Natl Acad Sci USA 90: 4961, 1993 |
|
Godwin AK, Perez RP, Johnson SW et al: Growth regulation of ovarian cancer. Ovarian Cancer 6: 829, 1992 |
|
Godwin AK, Testa JR, Handel LM et al: Spontaneous transformation of rat ovarian surface epithelial cells implicates repeated ovulation in ovarian cancer etiology and is associated with clonal cytogenetic changes. J Natl Cancer Inst 84: 592, 1992 |