Genital Duct Anomalies
Authors
INTRODUCTION
Abnormalities of the müllerian ducts produce a wide range of gynecologic and urologic disorders. The clinician must manage these problems and be prepared to counsel affected individuals concerning the recurrence risks to other family members. In this chapter, we consider the cause and pathogenesis of müllerian duct anomalies. Techniques for surgically correcting these anomalies are discussed elsewhere in these volumes and in other texts.1
DEVELOPMENT OF THE GENITAL TRACT
Sex begins at conception. The zygote contains the genetic material – 46,XX or 46,XY – that determines its future sex; however, male and female embryos are morphologically indistinguishable until 7 weeks of embryogenesis, when the male gonads first become distinct. The reproductive duct systems remain sexually indifferent until 12 weeks of gestation (10 weeks' embryogenesis). The embryo initially has two sets of paired ducts. The first are the mesonephric (wolffian) ducts, which in the male develop into the vas deferens, seminal vesicles, and epididymides. The second are the paramesonephric (müllerian) ducts, which in the female develop into the fallopian tubes, uterus, and upper vagina. Both sexes retain embryologic remnants of the duct that regress in their sex. Because perturbations of embryogenesis are responsible for the disorders discussed later in this chapter, a review of normal reproductive embryology seems appropriate.2, 3, 4
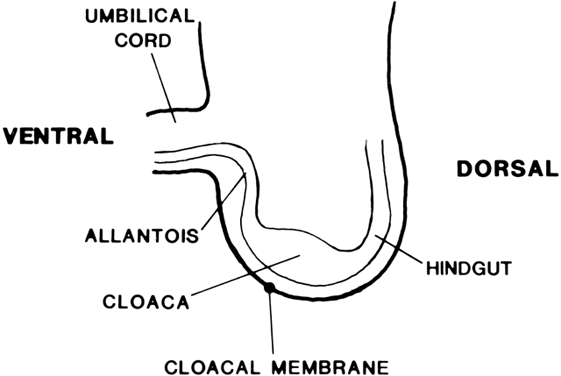
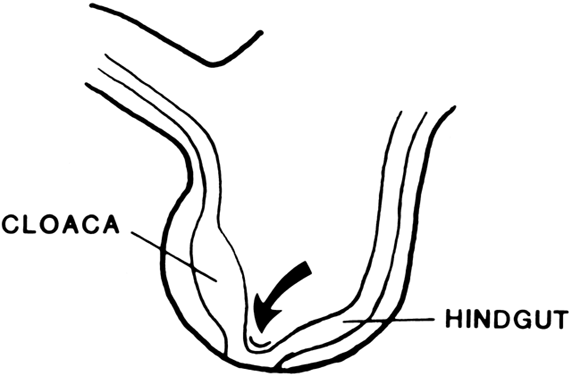
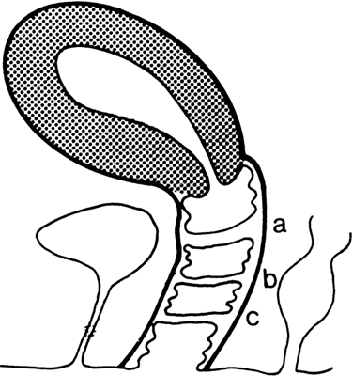
The undifferentiated embryo at 3 embryonic weeks contains a hindgut, a tube that runs from the dorsal aspect of what later will become the pelvic cavity, along the middle of the inferior pole, up the midline of the ventral embryo, and out the umbilical cord. The hindgut dilates as it passes along the inferior pole of the embryo, and at this point it is called the cloaca. The tube narrows again as it runs up the ventral embryo and here is called the allantois (Fig. 1). Between 4 and 6 embryonic weeks, a solid sheet of cells, the urorectal fold, grows downward to the cloacal membrane, indenting the hindgut–cloaca allantois tube and separating the cloaca from the hindgut (Fig. 2). The hindgut later develops into the sigmoid colon and rectum. Shortly after the urorectal fold meets it, the cloacal membrane breaks down and allows the urogenital sinus to open separately from the rectum.
{kind=link}
{kind=link}
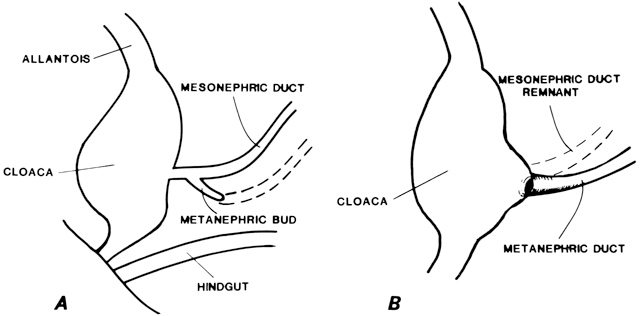
At 4 weeks of embryogenesis, tubes extend bilaterally from the embryonic mesonephros to the cloaca. These mesonephric ducts later become the vas deferens and epididymis in the male, but in the female, they eventually regress. Tubular buds arising from the dorsal surface of the mesonephric ducts grow upward to meet the metanephros, becoming the metanephric ducts (Fig. 3). The portion of mesonephric duct between the metanephric duct and cloaca dilates and becomes incorporated into the cloacal wall (Fig. 3) and later contributes to the formation of the bladder trigone. The metanephric ducts form the ureters.
{kind=link}
Embryonic development of males and females is similar. Subsequently, however, divergence occurs. Differential development of the male and female tubules is directed by the gonads. The principle is that all embryos develop female internal genitalia unless a functioning testis is present. Sertoli cells in the fetal testes produce müllerian-inhibiting factor (MIF), a glycoprotein that inhibits development of the paramesonephric (müllerian) ducts. Leydig cells in the fetal testes produce testosterone, which stabilizes the mesonephric (wolffian) ducts and promotes further development of vasa deferentia, epididymides, and seminal vesicles. Genital virilization is accomplished by dihydrotestosterone, which is converted from testosterone by 5α-reductase.
After 37 postovulatory days, celomic epithelium invaginates into the tissue lateral and cranial to the mesonephric duct. This solid mass of tissue grows caudally along the length of the mesonephric duct. Near the mesonephric–metanephric junctures, solid cords of tissue grow medially on both sides of the embryo toward the midline to fuse with each other. As the cords are growing caudally, a lumen appears in their cranial portion, extending toward the growing tip. These cords become the paramesonephric (müllerian) ducts, which fuse with the dorsal wall of the urogenital sinus to produce an elevation, the müllerian tubercle. In the presence of MIF these ducts do not develop further. In the absence of MIF (i.e. in normal females), paramesonephric ducts differentiate into fallopian tubes, uterus, and superior vagina.
Proliferation of the müllerian tubercle increases the distance between the urogenital sinus and the uterovaginal lumen. Concomitant with this elongation, tissue lateral to the fused paramesonephric ducts and lying at the base of the urogenital sinus proliferates to form sinovaginal bulbs. The sinovaginal bulbs grow caudally toward the urogenital diaphragm. When canalized, these bulbs form the vestibule of the vagina. The vagina is formed caudally from the sinovaginal bulbs of the urogenital sinus and cranially from the fused paramesonephric ducts. The precise proportion of vagina derived from each embryologic structure is uncertain, although most investigators believe the point of juncture lies at or slightly above the hymeneal ring. Most of the vagina is derived from the paramesonephric ducts. However, near-normal vaginal length in most patients with müllerian aplasia attests to the uncertainty or variable embryologic derivation.
Early fusion of the paramesonephric ducts is incomplete, with a septum persisting in the early uterus. By 9 embryologic weeks, the septum is no longer present; the uterocervical junction has developed. In the female, the mesonephric ducts by this time have begun to degenerate. Remnants of the mesonephric ducts persist in the broad ligament in females and are called Gartner ducts. The upper urogenital sinus develops into the bladder and the lower portion becomes the urethra.
ANATOMIC ANOMALIES
Vaginal atresia
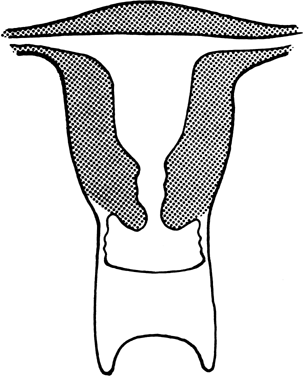
The vagina is shortened or absent in many females whose external genitalia are ambiguous (pseudohermaphrodites), but in the present context, we consider only those females who lack a vagina and whose external genitalia are otherwise normal. Two groups of individuals fulfill these characteristics: those with absence of most of the vagina and all or almost all of the uterus (müllerian aplasia) and those with absence of a portion of the vagina but presence of a normal uterus (vaginal atresia). The two conditions are embryologically and anatomically distinct (Fig. 4). Of individuals with an absent vagina, 80–90% have müllerian aplasia; the remainder have vaginal atresia.5, 6, 7, 8, 9, 10
{kind=link}
In vaginal atresia the urogenital sinus fails to contribute the caudal portion of the vagina. The lower fifth to third of the vagina is replaced by 2–3 cm of fibrous tissue, above which lie a well-differentiated upper vagina, cervix, uterine corpus, and fallopian tubes (Fig. 4). Ultrasound, magnetic resonance imaging, or rectal examination may verify presence of müllerian derivatives. Hydrometrocolpos can develop.
Familial aggregates of isolated vaginal atresia are rare if not nonexistent. However, vaginal atresia is often reported as part of a large series of patients with absence of vagina. Analysis of a heterogeneous sample of patients might obscure findings that would be evident if the two disorders were analyzed separately.
Vaginal atresia in multiple malformation syndromes
Etiologically distinct from vaginal atresia in otherwise normal women is vaginal atresia present as one component of a multiple malformation complex. Table 1 summarizes several syndromes. Winter and co-workers described four siblings with a previously unrecognized autosomal recessive syndrome characterized by vaginal atresia, renal hypoplasia or agenesis, and middle ear anomalies (malformed incus, fixation of the malleus and incus).11 A second malformation syndrome in which vaginal atresia occurs is the Fraser syndrome, characterized by vaginal atresia and by cryptophthalmus with its resultant blindness.12 Other syndromes include Antley-Bixler and Bardet-Biedl (Table 1).
Table 1. Multiple malformation syndromes associated with vaginal atresia
Syndrome | Somatic anomalies | Etiology |
| Antley-Bixler | Craniosynostosis, choanal atresia, humeroradial synostosis, gracile ribs, bowed femora, camptodactyly, renal anomalies | Autosomal recessive |
| Bardet-Biedl | Degeneration of retinal pigment (retinitis pigmentosa), polydactyly, obesity, mental retardation | Autosomal recessive |
| Fraser | Cryptophthalmia, nose and ear anomalies, stenotic larynx, skeletal defects, syndactyly, renal agenesis, large clitoris and labia majora, mental retardation | Autosomal recessive |
| Winter | Lacrimal duct stenosis, external and middle ear anomalies, renal agenesis | Autosomal recessive |
Transverse vaginal septa and McKusick-Kaufman syndrome
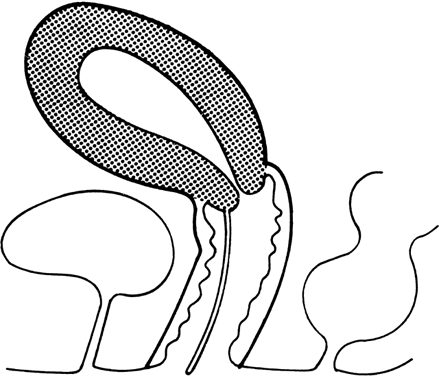
Transverse vaginal septa occur at several locations and may be complete or incomplete. These septa are usually about 2 cm thick and located near the junction of the upper third and lower two-thirds of the vagina;1, 2, 13 however, septa may be present in the middle or lower third of the vagina13 (Fig. 5). Perforations are usually central but may be eccentric in location.14, 15, 16, 17 If no perforation exists, mucus and menstrual fluid cannot egress; hydrocolpos or hydrometrocolpos may develop. Other pelvic organs are usually normal, although occasionally the uterus is bicornuate.
{kind=link}
Vaginal septa presumably result from failure of urogenital sinus derivatives and the müllerian duct derivatives to fuse or canalize. This explanation is deduced from the location of the septa, which is usually at the predicted sites of urogenital sinus müllerian fusion, and the histologic nature of the septa. The cranial surfaces of septa are usually lined by columnar (müllerian) epithelium, whereas caudal surfaces are lined by squamous epithelium (i.e. urogenital sinus invagination).
Some patients with transverse vaginal septa also have polydactyly, and cardiac defects.18 The original description and most subsequent cases have been in the Amish. The eponym McKusick-Kaufman syndrome is applied to such subjects.18 The latter cases indicate a pleiotropic mutant, a suggestion to which Pinsky subscribes.19 Alternatively, presence of multiple abnormalities may indicate a different mutant gene. Familial aggregates are rarely observed in non-Amish kindreds; it is difficult to distinguish between these possibilities.
In support of the thesis of a single pleiotropic gene is the analysis of 54 cases by Chitayat and colleagues.20 Hydrometrocolpos was estimated to be present in 95% of Amish cases, polydactyly in 93%, and cardiovascular malformations in 9%. Amish individuals may show all three anomalies, various pairwise combinations of two, or only one.21 Stone and colleagues estimated penetrance to be 70% for hydrometrocolpos in females, 60% for polydactyly in both sexes, and 15% for cardiovascular defects.22 Given these probabilities, 9% of males and 3% of females could be expected to have the gene in completely nonpenetrant state.
The MKS locus is on 20p12. There are six exons and an open reading frame of 570 amino acids beginning in exon 3. The protein is a chaperonin, representative of the class of proteins that facilitate protein folding in conjunction with adenosine triphosphate hydrolysis.23 Old Order Amish show two unusual chaperonin sequences (H84Y and A242S). Each variant is found in 1 per 100 Amish controls. H84Y/A242S compound heterozygosity segregates with the disorder. The 1% frequency of these variants in the general Amish population is consistent with a MKS heterozygote frequency of that magnitude. Compound heterozygotes are not necessarily clinically abnormal, meaning the heterozygote frequency of these variants is higher than 1% albeit to an unknown extent.
Vaginal longitudinal septa
Vaginal septa may be longitudinal (sagittal or coronal) (Fig. 6) or transverse. Longitudinal septa, which rarely produce clinical problems, probably result from abnormal mesodermal proliferation or persisting epithelium. Occasionally, these septa impede the second stage of labor. Heritable tendencies are not obvious, although no systematic studies have been reported.
{kind=link}
Edwards and Gale reported an autosomal dominant syndrome characterized by longitudinal vaginal septum, hand anomalies, and urinary incontinence possibly because of a bladder neck anomaly.24 Longitudinal vaginal septa also occurs in the Johanson-Blizzard syndrome, which is probably an autosomal recessive disorder25 (Table 2).
Table 2. Syndromes associated with longitudinal vaginal septa
Syndrome | Somatic anomalies | Etiology |
| Edwards-Gale (camptobrachydactyly)24 | Flexion contractures of distal interphalangeal joints, brachydactyly, polydactyly, syndactyly, urinary incontinence | Autosomal dominant |
| Johanson-Blizzard25 | Scalp defects, deafness, hypoplastic alae nasi, microdontia, primary hypothyroidism, malabsorption, mental retardation, hypotonia, short stature | Autosomal recessive |
Absence or atresia of the uterine cervix
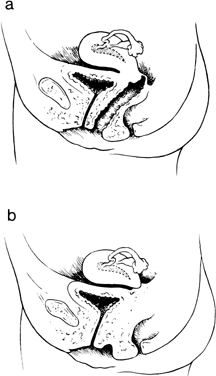
Isolated absence or hypoplasia of the cervix associated with a normal uterine corpus and a normal vagina is rare. Relatively few cases have been described, and there have been no reports of multiple affected family members.26, 27 The disorder presumably results from failure of müllerian duct canalization or increased local epithelial proliferation after canalization. Hydrometrocolpos should be anticipated. The cervical canal may also be absent in true hermaphrodites.28
In 1997, Fujimoto and colleagues reported seven new cases and reviewed the 51 previously reported cases.29 They concluded that one half of all cases with cervical absence or atresia had normal vaginas; one half had complete or partial vaginal atresia (Fig. 7). Surgically created uterovaginal canalization led to menstruation in 60% of cases overall, but more often if concomitant vaginoplasty was not concurrently needed (68% versus 43%). After surgical correction, pregnancies have occurred only exceptionally.27, 28, 29, 30
{kind=link}
Müllerian aplasia
Aplasia of the müllerian ducts leads to absence of the uterine corpus, the uterine cervix, and the upper portion of the vagina (Fig. 4). The foreshortened 1–2 cm vagina is presumably derived exclusively from invagination of the urogenital sinus. Individuals with müllerian aplasia usually consult physicians because of primary amenorrhea. Secondary sexual development is normal, but no uterine structures are palpable. Uterine remnants may exist in the form of bilateral cords. The term Rokitansky-Küster-Hauser syndrome, is often applied, sometimes if remnants persist and sometimes synonymously with müllerian aplasia.
The only disorder that ordinarily needs to be considered in the differential diagnosis is complete androgen insensitivity. Androgen insensitivity can be excluded on the basis of chromosomal studies and gonadal composition. Pubertal patients with müllerian aplasia invariably have pubic hair, whereas those with androgen insensitivity usually do not.
Renal anomalies are associated with müllerian aplasia more frequently than would be expected by chance (Table 3).9, 31, 32, 33, 34 The most frequent renal anomalies are pelvic kidney, renal ectopia, and unilateral aplasia. Skeletal anomalies, especially vertebral anomalies, are not uncommon. Excretory urography and vertebral roentgenograms are obligatory in the clinical evaluation of müllerian aplasia.
Table 3. Urologic anomalies in müllerian aplasia
Study | Total sample | No. undergoing urography | No. with renal anomaly |
Phelan et al.31 | 129 | 72 | 26/72 (8 “minor”) |
Thompson et al.32 | 17 | 17 | 8/17 |
Leduc et al.9 | 10 | 10 | 4/10 |
Fore et al.33 | 32 | 32 | 20/32 |
Carson et al.34 | 23 | 22 | 4/22 (18.2%) |
Total | 211 | 153 | 62/153 (40.5%) |
Familial aggregates of müllerian aplasia have been reported, namely affected siblings.35, 36, 37, 38, 39 However, Lischke and associates observed three sets of discordant monozygotic twins, and autosomal recessive inheritance therefore is an unlikely explanation for all cases.40
Autosomal dominant inheritance was considered by Shokeir to exist in Saskatchewan families in which the proband had müllerian aplasia.39 In 13 of 16 families, the proband showed complete absence of the uterine cervix and corpus; in the remaining three, uterine remnants (Rokitansky-Küster-Hauser) were present. None of the three individuals with uterine remnants had an affected relative, but 10 of the 13 with complete absence of the uterine cervix and corpus did. Two of these 10 had affected siblings, whereas the other eight had other affected paternal relatives (i.e. aunts, first cousins, second cousins, or great-aunts). Such observations suggest sex-limited (female) autosomal dominant inheritance, although other genetic mechanisms cannot be excluded. Females with the postulated mutant would manifest müllerian abnormalities, whereas males would show no deleterious effect. Lack of confirmatory reports over the subsequent 30 years still leaves the significance of the observations of Shokeir39 unclear.
In contrast, Carson et al.34 reported 23 US families in which not a single relative was affected. Absence of affected relatives among 30 postpubertal sisters, 31 paternal aunts, and 41 maternal aunts makes sex-limited autosomal dominant inheritance at least uncommon; however, dominant genes could be restricted to certain populations, and fresh dominant mutations can never be excluded. However, absence of affected siblings and lack of paternal consanguinity speaks against autosomal recessive inheritance. Van Lingen et al.41 reported one set of siblings among 35 cases.
Women with müllerian aplasia have normal ovaries. Given this, a therapeutic strategy is to obtain oocytes from affected women, perform fertilization in vitro with their husband's sperm, and transfer fertilized embryos to the surrogate uterus of another woman in hormonal synchrony. The resulting offspring would genetically reflect the affected woman. Petrozza and colleagues42 surveyed US assisted reproductive technology (ART) programs to accumulate 34 pregnancies in women with müllerian aplasia. Of the 34 offspring, 17 were female, and none were affected; one male child had a middle ear defect and hearing loss.
These data are most consistent with polygenic/multifactorial inheritance, the usual mode of inheritance for malformations affecting a single organ system or embryologically related systems. Müllerian aplasia clearly fulfills these characteristics. Polygenic/multifactorial inheritance could explain the occasional reports of multiple affected siblings. After the birth of one child with a polygenic/multifactorial disorder, the recurrence risk for first-degree relatives of affected probands approximates the square root of the incidence of the trait in the population. Because müllerian aplasia is rare, the recurrence risk for siblings should be low. A theoretical recurrence risk could be calculated if accurate incidence data existed. (Risk equals the square root of incidence of the trait.) Failure to detect affected sibs in a relatively small sample is consistent with polygenic/multifactorial inheritance and a low (1–2%) recurrence risk for first-degree relatives.
The other plausible explanation is genetic (etiologic) heterogeneity. A dominant or recessive gene could explain a minority of cases, perhaps only in certain populations; nongenetic factors or polygenic/multifactorial inheritance could explain the remainder.
Little progress has been reported in the molecular elucidation of müllerian aplasia. Using denatured gradient gel electrophoresis (DGGE), various studies in the 1990s sought and found no molecular abnormalities in a number of candidate genes. However, these studies could exclude only large deletions. Studying WT143 and PAX241 no perturbations were found. This also held for AMH and AMHR.44 The N314 allele of GALT45, 44 was not increased. Perturbations of HOXA13 cause the autosomal dominant condition hand-foot-genital syndrome, but HOXA13 mutations have not been found in isolated müllerian aplasia. More recent and more detailed studies (sequencing) include those of Cheroki et al.,46 who in 25 cases excluded WNT4, RAR-Gamma and RXR-alpha perturbations. Burel47 found no perturbations in Hox genes (A7-A13) or PBX in a sample of six cases. Ma et al.48 found no perturbations in sequencing 192 Han Chinese cases.
Sultan et al.49 studied 28 cases for mutation in WNT4, finding one heterozygous missense mutation (L12P).
All these studies involve very small sample sizes and all involve only European populations. Several groups have performed array comparative genomic hybridization (CGH)50, 51, 46, 52, 53, 54, 55 showing a variety of microdeletions and microduplications (i.e., less than the 5 Mb required for recognition by high resolution CTG-banding studies). These include 1q21.1, 16p11.2, 17q12, and 22q11.2, all common variants. Familial transmission likely indicative of benign polymorphisms was not usually excluded, unfortunately because these regions are among those most commonly detected in array CGH studies irrespective of the indication for performing CGH (e.g., postnatal infant with development delay, fetus with ultrasound detected anomalies). Del22q11.2 is probably most likely to play a role in certain cases, whether de novo or familial (if paternal). Microdeletions and microduplications of these regions are also found in normal individuals; thus, ascribing a causative role in müllerian aplasia is hazardous. A illustrative study receiving attention is that of Gervasini56 who, in a study of 30 müllerian aplasia cases, found a duplication of Xq21 only in five müllerian aplasia cases (two sporadic, three familial). Especially intriguing was a family of two affected müllerian aplasia sibs who received a 17 kb duplication from their father. Two sibs having a uterus failed to inherit the microduplication. The pseudoautosomal region duplicated contained SHOX, a homeobox gene that escapes X-inactivation and has been related to short stature if deficient and to Leri-Weill syndrome and Langer mesomelic dysplasia if duplicated. 57, 58 However, array CGH findings in müllerian aplasia were not confirmed in the much more robust study of Sandbacka et al.59 of 101 cases. This study noted that all copy number variations reported by Gervasini are in fact recorded in the Database of Genomic Variants and considered without phenotypic effect.
A molecular perturbation has been found in müllerian aplasia only in a very atypical case. Biason-Lauber et al.60 found perturbations of ANT4 in many typical cases of müllerian aplasia in which virilization and adrenal insufficiency existed. The relevance to isolated müllerian aplasia is unclear. Gervasini et al.56 found no perturbations in 12 müllerian aplasia cases who also had hyperandrogenism. WNT4, acts before AMH and is required for initial müllerian development,61however, so plausibility could exist.
In several cases report women with müllerian aplasia with a de novo balanced translocation. Kucheria and co-workers62 reported two unrelated Indian females with 46,XX t(12;14)(q14;q31); no information was given on whether this ostensibly balanced translocation was familial. This observation is nonetheless intriguing because AMHR is localized to 12q13. However, no AMH or AMHR perturbations were found in müllerian aplasia by Resendes and colleagues.63 Van Lingen and co-workers64 reported a de novo t(3;6)(p23;13.3); polymorphic DNA markers near these break points showed no perturbations. A de novo t(8;13)(q22.1;q32.1) translocation was reported by Ammese and co-workers.65
Müllerian aplasia and multiple malformation syndromes
Renal and vertebral differentiation are embryologically related, but a few patients display the unexpected combination of müllerian aplasia and fusion of cervical vertebrae (Klippel-Feil anomaly). Coexistence of müllerian aplasia and Klippel-Feil anomaly is sometimes associated with middle ear anomalies.9, 66, 67, 68, 69, 70 This triad could indicate an entity distinct from more common forms of müllerian aplasia, especially given that renal anomalies were not present in individuals with Klippel-Feil anomaly. Neurosensory hearing loss in the high-frequency range has been observed.71
In several other multiple malformation syndromes, müllerian aplasia is one component (Table 4). The mechanisms presumably reflect perturbation of genes different from those responsible for müllerian aplasia in otherwise normal individuals.
Table 4. Syndromes associated with müllerian aplasia
Syndrome | Somatic anomalies | Etiology |
| Fraser | Cryptophthalmia, nose and external ear anomalies, stenotic larynx, skeletal defects, syndactyly, renal agenesis, large clitoris and labia majora, mental retardation | Autosomal recessive |
| Meckel-Gruber | Microcephaly, posterior encephalocele, eye anomalies, cleft palate, polydactyly, polycystic kidneys | Autosomal recessive |
| MURCS association | Renal aplasia, cervicothoracic somite dysplasia, Klippel-Feil anomaly, deafness, short stature | Unknown |
| Thalidomide teratogenicity | Nasal hemangioma, neurosensory hearing loss, ear anomalies, limb reduction defects, visceral anomalies | Teratogen |
| Urogenital adysplasia (hereditary renal adysplasia; bilateral, renal agenesis) | Oligohydramnios, flattened (Potter) facies, pulmonary hypoplasia, unilateral or bilateral absent kidneys, limb deformities | Autosomal dominant |
| Winter | Lacrimal duct stenosis, external and middle ear anomalies, renal agenesis | Autosomal recessive |
True duplication of the müllerian ducts
True duplication of the uterus is a rare anomaly that probably results from division of one or both müllerian ducts early in embryogenesis. Affected individuals have two separate uteri, each of which may have two fallopian tubes.2 One or both uteri may be rudimentary or bicornuate. True duplication should be distinguished from incomplete müllerian fusion, the much more frequent condition in which each of two hemiuteri is associated with only a single fallopian tube. Understandably because hemiuteri are so much more common than true duplications, the frequent practice of referring to bicornuate uteri as a “double uterus” actually constitutes a misnomer. No familial aggregates have been reported.
Incomplete müllerian fusion
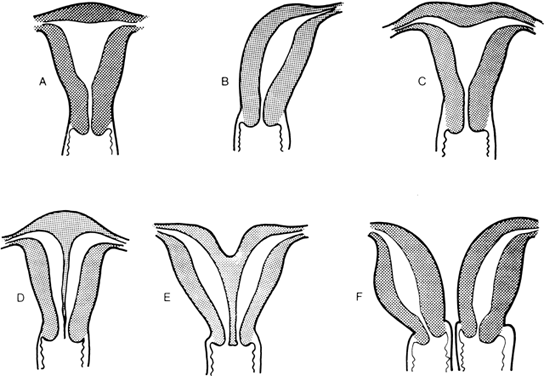
The müllerian ducts are originally paired organs that fuse and canalize to form the upper vagina, uterus, and fallopian tubes. Failure of fusion results in two hemiuteri, each associated with no more than one fallopian tube. Sometimes one müllerian duct fails to contribute to the definitive uterus, leading to a rudimentary horn. Fig. 8 shows the different varieties of incomplete müllerian fusion. Renal anomalies coexist with all. If one uterine horn is atretic, ipsilateral renal agenesis is especially common.
{kind=link}
Familial aggregates of incomplete müllerian fusion include multiple affected siblings and affected mothers and daughters.72, 73, 74, 75, 76, 77, 78, 79, 80 Individuals in the same kindred may show different forms of incomplete müllerian fusion.80 In the only formal genetic study reported,81 only one (2.7%) of 37 sisters had a clinically symptomatic uterine anomaly. There were no affected mothers (0 of 24), maternal aunts (0 of 44), or paternal aunts (0 of 50). The 2.7% prevalence in siblings constitutes a minimum frequency because relatives could have had a minor uterine anomaly in asymptomatic form. Ideally, hysteroscopy, hysterosalpingography, or surgical exploration could be performed on relatives. Female relatives in some families had not yet attempted pregnancy, limiting the opportunity to manifest symptoms that would suggest an anomaly. Even with such inherent limitations, the likelihood of first-degree female relatives being similarly affected with müllerian fusion anomalies would seem to be too low to be compatible with an autosomal dominant or autosomal recessive origin. That approximately 3% of female siblings were affected in the one formal study is consistent with predictions based on polygenic/multifactorial cause, assuming further studies confirm the previously given data. The only molecular study reported in that of Ma et al.48 who found no mutations in 192 cases.
Hand-foot-genital syndrome
The hand-foot-genital (HFG) syndrome is an autosomal dominant disorder in which incomplete müllerian fusion is a major component. First reported by Stern and associates, multiple kindreds have now been recognized.82, 83, 84 A family first identified by our group75 was updated by Donnenfeld and colleagues.85 The syndrome is characterized by skeletal (hand and foot) malformations and incomplete müllerian fusion in females or hypospadias in males (with “hand-foot-genital” replacing the original appellation of “hand-foot-uterus”).85, 86 Limb abnormalities include short first metacarpals, small distal phalanges on the thumbs, short middle phalanges on the small finger and fusion of the wrist bones. Analogously, the great toe is short because of a shortened metatarsal, and the phalanx is small and pointed.
Urinary system anomalies include urinary incontinence (female), ventral displaced urethral meatus (male and female), and malposition of the ureteral orifices in the bladder wall (female).87 These urologic anomalies differ from those usually associated with incomplete müllerian fusion. Vertebral anomalies do not seem characteristic of the HFG syndrome.
HFG syndrome should be sought in all females with uterine anomalies, because offspring of affected women have a 50% likelihood of inheriting the mutant gene. Inquiry should be made concerning the presence or absence of skeletal anomalies or genital anomalies in male and female relatives. Even in the absence of a positive family history, HFG syndrome may be considered to be present if an individual displays characteristic skeletal and genital anomalies. Such an individual would probably represent a new mutation. It is also relevant to recall that varied expressivity is characteristic of all autosomal dominant disorders. It is possible that some females with the HFG gene may manifest only uterine anomalies or only skeletal anomalies, whereas others in the same kindred show both.
That the skeletal anomalies in HFG syndrome were reminiscent of the hypodactyly (Hd) mutant in the mouse was recognized by Mortlock and colleagues,88 who had earlier detected a deletion in murine exon 1 of HOXA13.89HOXA13 was a good candidate gene for human HFG. A HOXA13 nonsense mutant was observed in a member of the original HFG family reported by Stern and co-workers.83 In one of the HFG cases studied by Goodman et al.,90 a HOXA13 mutation also involved a stop codon, and again led to a truncated protein. In another family, a HOXA13 mutation involved expansion of the polyadenosine (poly A) tail, suggesting a dominant negative mechanism. The manner by which perturbation of HOXA13 produces HFG is still uncertain, but HOXA13 is integral for differentiation or fusion/canalization of müllerian derivatives.
Incomplete müllerian fusion and other multiple malformation syndromes
HFG is not the only one multiple malformation syndrome associated with incomplete müllerian fusion. Table 5 shows a more complete list. Many different genes and nonmendelian factors must remain intact for normal uterine development. Whether wild-type genes for these syndromes are integral for normal müllerian differentiation is unclear. In some of these syndromes uterine anomalies may arise secondary to connective tissue or vascular perturbations. The wild-type genes are not part of the normal müllerian differentiation cascade.
Table 5. Syndromes associated with incomplete müllerian fusion
Syndrome | Somatic anomalies | Etiology |
Bardet-Biedl | Retinal pigmentary degeneration (retinitis pigmentosa), polydactyly, obesity, mental deficiency polydactyly, obesity, mental deficiency | Autosomal recessive |
Beckwith-Wiedemann | Macroglossia, omphalocele, macrosomia | Autosomal dominant, after uniparental disomy |
Donohue (leprechaunism) | Elfin facies with thick lips; large, low-set ears; prominent breasts and external genitalia; hirsutism; abnormal carbohydrate metabolism; failure to thrive; motor and mental retardation | Autosomal recessive |
Fraser | Cryptophthalmia, external ear and nose anomalies, laryngeal stenosis, syndactyly, skeletal defects, renal agenesis, large clitoris and labia majora, mental retardation | Autosomal recessive |
Hand-foot-genital (HFG) | Metacarpal and metatarsal anomalies, malformed thumbs, displaced urethral meatus, urinary incontinence | Autosomal dominant |
Johanson-Blizzard | Deafness, hypoplastic alae nasi, primary hypothyroidism, mental retardation | Autosomal recessive |
Laryngeal atresia | Hydrocephaly, complete or partial laryngeal obstruction, trachesophageal fistula or atresia, renal hypoplasia, varus deformity of feet | Unknown |
Meckel-Gruber | Microcephaly, posterior encephalocele, eye anomalies, cleft palate, polycystic kidneys, polydactyly | Autosomal recessive |
Roberts | Sparse, silvery blond hair; midfacial hemangioma; cleft lip with or without cleft palate; limb reduction defect; intrauterine growth retardation | Autosomal recessive |
Rudiger | Bifid uvula, coarse facies, absent ear cartilage, hydronephrosis secondary to ureterovesical stenosis, short digits | Autosomal recessive |
Thalidomide teratogenicity | Nasal hemangioma, neurosensory hearing loss, ear anomalies, limb reduction defects, visceral anomalies | Teratogen |
Trisomy 18 | Prominent occiput, malformed ears, micrognathia, short sternum, cardiac defects, horseshoe kidney, overlapping fingers, intrauterine growth retardation, severe developmental retardation | Chromosomal aneuploidy |
Trisomy 13 | Microcephaly, microphthalmia, malformed ears, cleft lip and palate, cardiac anomalies, polydactyly, intrauterine growth retardation, severe developmental retardation | Chromosomal aneuploidy |
Urogenital adysplasia, hereditary (hereditary renal agenesis) | Oligohydramnios, flattened (Potter) facies, pulmonary hypoplasia, unilateral or bilateral absent kidneys, limb deformities | Autosomal dominant |
Imperforate hymen
Ordinarily, the central portion of the hymen is patent (perforate), allowing outflow of mucus and blood. If the hymen is imperforate, mucus and blood accumulate in the vagina or uterus (i.e. hydrocolpos or hydrometrocolpos). An imperforate hymen is not uncommon. Fortunately, the anomaly is easily corrected by surgical incisions, preferably cruciform. McIlroy and Ward91 and Usta et al.92 have both reported siblings who possibly had the disorder. Stelling and colleagues93 reported concordant monozygotic twins; one twin had an affected daughter, who also had pyloric stenosis and possibly hip dislocation.
Isolated absence of fallopian tubes
Absence of a fallopian tube in an otherwise normal female is rare.2, 94 Fallopian tubes usually persist despite regression of all other müllerian derivatives (i.e. uterus, cervix, and upper vagina). Unilateral absence of the ovary may accompany ipsilateral absence of the fallopian tube.76 This implies pathogenesis involves a vascular accident or torsion after completion of gonadal and ductal differentiation, perhaps analogous to the cause of anorchia.95 No familial aggregates have been reported.
Persistence of müllerian derivatives in males
The uterus and fallopian tubes (müllerian derivatives) may persist in ostensibly normal (46,XY) males. External genitalia, wolffian (mesonephric) derivatives, and testes develop as expected for males, and pubertal virilization occurs.
Testes are presumed to differentiate normally, but they may not remain normal. One or more testes may be present in the intra-abdominal or inguinal region, and undergo secondary degeneration. About 5% of individuals develop a seminoma or other germ cell tumor. Testes in persistent müllerian derivatives (PMDs) are also abnormally mobile96 as result of not being anchored properly to the processus vaginalis. Increased mobility could predispose to testicular torsion and secondary testicular degeneration, which are frequent in PMD.97 Infertility is common for these and perhaps other reasons.
Two genes are integral for precluding müllerian development in men. One gene codes for antimüllerian hormone (AMH; formerly called MIS); the other codes for the AMH receptor (AMHR). Josso and colleagues have studied AMH and AMHR in over 100 PMD families, in 15% no mutation was identified.98 Mutations are equally likely to involve AMH or the AMH receptor (AMHR). In a 2005 report, 82 cases of PMD in men were analyzed by Josso and co-workers.99 Of these, 38 showed an AMH mutation, 33 an AMH receptor mutation (AMHR), and 11 neither.99
Antimüllerian hormone mutation
Located on 19p13.3, AMH consists of five exons. The 3′ end is guanine cytosine (GC) rich. The AMH gene product can be measured by enzyme-linked immunosorbent assay, but results are informative only before sexual maturation. AMH production is suppressed after puberty. When AMH is not detected, a mutation in the structural gene can usually be demonstrated. Although Imbeaud and co-workers100 initially failed to detect recurrent AMH mutations in 19 PMD families (molecular heterogeneity), recurrent mutations were later found.101 In a 2005 tabulation, 38 different mutations were found in 46% of families studied.99 Most mutations were homozygous and found in individuals of North African (Arab) or Mediterranean descent.
AMH receptor
The AMH receptor (AMHR II) gene is located on 12q13.12. It consists of 11 exons and is 8700 bp in length. AMHR II mutations have now been found throughout the gene. Compared with AMH-negative cases, AMH-positive cases are relatively less likely to be found in North African Arab populations.
If AMH is elevated, an AMHR mutation should be suspected. Most mutations found are missense, generally occurring throughout the gene. Mutations in exon 1 and in the 3′ portion of exon 5 are most common. The most frequent single perturbation is deletion of 27 base pairs in exon 10 (del 6331-6357). When present, the deletion exists in homozygous form in 42% of cases; compound heterozygosity exists in 58%, the deletion coupled with a missense mutation.102 For diagnosis, Belville and co-workers101 recommend polymerase chain reaction (PCR) to detect the 27 bp mutation, followed by sequencing the entire AMHR gene if the deletion is not present in homozygous form.
Women (46,XX) with AMHR mutations undergo puberty normally.102 Actually this is a mild surprise because transgenic mice that chronically express AMH show gonadal abnormalities.103 Granulosa cells also normally produce AMH104, 105 for which reason a role for AMH in gonadal development seems plausible.
Congenital absence of the vas deferens and cystic fibrosis
Almost all men with cystic fibrosis (CF) are infertile, specifically as result of congenital absence of the vas deferens (CBAVD). At least 70%, if not all men with CBVAD, have one of more mutations in CFTR. The most common CF mutations causing CAVD are ΔF508 and W128X. In addition to the missense and nonsense mutations in CFTR causing classic CF with pancreatic and pulmonary pathology, a deleterious polymorphism of specific relevance to CBAVD exists when 5-thymidines (5-T) are present in a particular location in intron 8.106 The 5T polymorphism (allele) results in very low (10%) transcription of CFTR from that chromosome (cis), owing to improper exon–intron splicing and subsequent loss of exon 9. The 7T polymorphism has less effect, and 9T none. If 5T "polymorphism" is homozygous, bilateral CAVD (CBAVD) results. Classic CP (pulmonary and pancreatic disease) does not result because the 10% CFTR gene product is sufficient for normal lung and pancreas function. ΔF508/5T compound heterozygosity leads to CBAVD. However, if 5T exists cis with R117H, that chromosome produces no CFTR. Thus ΔF508/R117H + 5T results in classical CF with pancreatic and pulmonary symptoms.
For the purpose of genetic counseling, CAVD cases should be assumed to involve compound heterozygosity for two mutant CF alleles, or one CF allele and the 5T variant. It should be assumed that both CF alleles are dysfunctional, even if one or both are not diagnostically evident. The exception arises if unilateral renal aplasia is present.107
Failure of fusion of epididymis and testis
Another relatively common urologic defect is failure of the testicular rete cords of the testis to fuse with the mesonephric tubules destined to form the ductule efferentia. Spermatozoa cannot exit and if the defect is bilateral, infertility results. One or both testes may also fail to descend.
Fusion defects of this type occur in about 1% of cryptorchid and in about 1% of azoospermic men. Familial aggregates have not been reported. Fertility is achievable by aspirating sperm from the testes or epididymis and using assisted reproductive technologies like intracytoplasmic sperm injection.
REFERENCES
Jones HW Jr, Rock JA: Reparative and Constructive Surgery of the Female Generative Tract. Baltimore: Williams & Wilkins, 1983 |
|
Simpson JL (ed): Disorders of Sexual Differentiation: Etiology and Clinical Delineation. New York: Academic Press, 1976 |
|
Pattern MP: Human Embryology, pp 419–499. 3rd ed. New York: McGraw-Hill, 1968 |
|
Hamilton WJ, Mossman HW: Human Embryology, pp 377–436. 4th ed. Baltimore: Williams & Wilkins, 1972 |
|
Bryan AL, Nigro JA, Counseller VS: One hundred cases of congenital absence of vagina. Surg Gynecol Obstet 88: 79, 1949 |
|
Cali RW, Pratt JH: Congenital absence of the vagina. Am J Obstet Gynecol 100: 752, 1968 |
|
Jones HW Jr, Scott WM: Hermaphroditism, Genital Anomalies, and Related Endocrine Disorders. 2nd ed. Baltimore: Williams & Wilkins, 1971 |
|
Jones HW Jr, Wheeless CR: Salvage of the reproductive potential of women with anomalous development of the muellerian ducts: 1868-1968-2068. Am J Obstet Gynecol 104:348, 1969 |
|
Leduc B, Campenhout J, Simard R: Congenital absence of the vagina. Am J Obstet Gynecol 100: 512, 1968 |
|
Turunen A, Unnerus CE: Spinal changes in patients with congenital aplasia of the vagina. Acta Obstet Gynecol 46: 99, 1967 |
|
Winter JSD, Kohn G, Mellman WJ et al: A familial syndrome of renal, genital and middle ear anomalies. J Pediatr 71: 88, 1968 |
|
Fraser GR: Our genetical “load”: A review of some aspects of genetic malformations. Ann Hum Genet 25: 387, 1962 |
|
Lodi A: Contributo clinico statistico sulle malformazioni della vagina osservate nella Clinica Obstetrica e Gineocologica de Milano del 1906 al 1950. Ann Obstet Ginecol 73: 1246, 1951 |
|
Bowman JA Jr, Scott RB: Transverse vaginal septum: Report of four cases. Obstet Gynecol 3: 441, 1954 |
|
Deppisch LM: Transverse vaginal septum. Obstet Gynecol 39: 193, 1972 |
|
Kanagasuntheran R, Dassanayake AGS: Nature of the obstructing membrane in primary cryptomenorrhea. J Obstet Gynecol Br Commonw 65: 487, 1958 |
|
White AJ: Vaginal atresia: High transverse septum. Obstet Gynecol 27: 695, 1966 |
|
Kaufman RL, Hartmann AF, McAlister WH: Family studies in congenital heart disease: II. A syndrome of hydrometrocolpos, postaxial polydactyly and congenital heart disease. Birth Defects 8: 85, 1972 |
|
Pinsky L: A community of human malformation syndromes involving the müllerian ducts, distal extremities, urinary tract, and ear. Teratology 9: 65, 1974 |
|
Chitayat D, Hahm SY, Marion RW et al: Further delineation of the McKusick-Kaufman hydrometrocolpos-polydactyly syndrome. Am J Dis Child 141: 1133, 1987 |
|
McKusick VA: The William Allen memorial award lecture: Genetic nosology: Three approaches. Am J Hum Genet 30: 105, 1978 |
|
Stone DL, Agarwala R, Schaffer AA et al: Genetic and physical mapping of the McKusick-Kaufman syndrome. Hum Mol Genet 7: 475, 1998 |
|
Stone DL, Slavotinek A, Bouffard GG et al: Mutation of a gene encoding a putative chaperonin causes McKusick-Kaufmansyndrome. Nat Genet. 2000 May;25(1):79-82. |
|
Edwards JA, Gale RP: Camptobrachydactyly: A new autosomal dominant trait with two probably homozygotes. Am J Hum Genet 24: 464, 1972 |
|
Johanson A, Blizzard R: A syndrome of congenital aplasia of the alae nasi, deafness, hypothyroidism, dwarfism, absent permanent teeth, and malabsorption. J Pediatr 79: 982, 1971 |
|
Geary WL, Weed JC: Congenital atresia of the uterine cervix. Obstet Gynecol 42: 213, 1973 |
|
Farber M, Marchant DJ: Reconstructive surgery for congenital atresia of the uterine cervix. Fertil Steril 27: 1277, 1976 |
|
Van Niekerk WA: True Hermaphroditism. New York: Harper & Row, 1974 |
|
Fujimoto VY, Miller JH, Klein NA et al: Congenital cervical atresia: Report of seven cases and review of the literature. Am J Obstet Gynecol 177: 1419, 1977 |
|
Fraser I: Successful pregnancy in a patient with congenital partial cervical atresia. Obstet Gynecol 74: 443, 1989 |
|
Phelan JT, Counseller VS, Greene LF: Deformities of the urinary tract with congenital absence of the vagina. Surg Gynecol Obstet 97: 1, 1953 |
|
Thompson JD, Wharton LR, Te Linde RW: Congenital absence of the vagina: An analysis of thirty-two cases corrected by the McIndoe operation. Am J Obstet Gynecol 74: 397, 1957 |
|
Fore SR, Hammond CB, Parker RT: Urologic and genital anomalies in patients with congenital absence of the vagina. Obstet Gynecol 46: 410, 1975 |
|
Carson SA, Simpson JL, Malinak LR et al: Heritable aspects of uterine anomalies: II. Genetic analysis of müllerian aplasia. Fertil Steril 40: 86, 1983 |
|
Kupperman HJ: Human Endocrinology, vol 1, p 261. Philadelphia: FA Davis, 1963 |
|
Anger D, Hamet J, Ensel J: Forme familiale du syndrome de Rokitansky-Kuster-Hauser. Bull Fed Soc Gynecol Obstet Lang Fr 18: 299, 1966 |
|
Jones HW Jr, Mermut S: Familial occurrence of congenital absence of the vagina. Am J Obstet Gynecol 114: 1100, 1972 |
|
Griffin JE, Edwards C, Madden JD et al: Congenital absence of the vagina, the Mayer-Rokitansky-Kuster-Hauser syndrome. Ann Intern Med 85: 224, 1976 |
|
Shokeir MHK: Aplasia of the müllerian septum: Evidence of probably sex-limited autosomal dominant inheritance. Birth Defects 14: 171, 1978 |
|
Lischke JH, Curtis CH, Lamb EJ: Discordance of vaginal agenesis in monozygotic twins. Obstet Gynecol 41: 920, 1973 |
|
van Lingen BL, Eccles MR, Reindollar RH, et al. (1998) Molecular genetic analysis of the PAX2 gene in patients with congenital absence of the uterus and vagina. Fertil Steril 70:S402. |
|
Petrozza JC, Gray MR, Davis AJ et al: Congenital absence of the uterus and vagina is not commonly transmitted as a dominant genetic trait: Outcomes of surrogate pregnancies. Fertil Steril 67: 387, 1997 |
|
van Lingen BL, Reindollar RH, Davis AJ et al: Further evidence that the WT1 gene does not have a role in the development of thederivatives of the mullerian duct. Am J Obstet Gynecol. 1998 Sep;179(3 Pt 1):597-603. |
|
Zenteno JC, Carranza-Lira S, Kofman-Alfaro S: Molecular analysis of the anti-Mullerian hormone, the anti-Mullerian hormone Arch Gynecol Obstet. 2004 May;269(4):270-3. |
|
Bhagavath B, Stelling JR, van Lingen BL et al. (1998) Congenital absence of the uterus and vagina (CAUV) is not associated with the N314D allele of the galacrose-1-phosphate uridyl transferase (GALT) gene. J Soc Cynecol Investig 5:140. |
|
Cheroki C, Krepischi-Santos AC, Rosenberg C et al: Report of a del22q11 in a patient with Mayer-Rokitansky-Kuster-Hauser (MRKH)determining MRKH anomaly in a study of 25 affected women. Am J Med Genet A. 2006 Jun 15;140(12):1339-42. |
|
Burel A, Mouchel T, Odent S et al: Role of HOXA7 to HOXA13 and PBX1 genes in various forms of MRKH syndrome(congenital absence of uterus and vagina). J Negat Results Biomed. 2006 Mar 23;5:4. |
|
Ma J, Qin Y, Liu W et al: Analysis of PBX1 mutations in 192 Chinese women with Mullerian ductabnormalities. Fertil Steril. 2011 Jun 30;95(8):2615-7. |
|
Sultan C. Lumbroso S (1998) LH receptor defects. In Kenpers RD, Cohen J. Haney AF, Younger JB (eds.); Fertility and Reproductive Medicine - Preceedings of the XVI World Congress on Fertility and Sterility. Elsevier Science, Amsterdam, pp.769-782. |
|
Bendavid C, Pasquier L, Watrin T et al: Phenotypic variability of a 4q34-->qter inherited deletion: MRKH syndrome in thedaughter, cardiac defect and Fallopian tube cancer in the mother. Eur J Med Genet. 2007 Jan-Feb;50(1):66-72. |
|
Bernardini L, Gimelli S, Gervasini C et al: Recurrent microdeletion at 17q12 as a cause of Mayer-Rokitansky-Kuster-Hauser(MRKH) syndrome: two case reports. Orphanet J Rare Dis. 2009 Nov 4;4:25. |
|
Cheroki C, Krepischi-Santos AC, Szuhai K et al: Genomic imbalances associated with mullerian aplasia. J Med Genet. 2008 Apr;45(4):228-32. |
|
Ledig S, Schippert C, Strick R et al: Recurrent aberrations identified by array-CGH in patients with Fertil Steril. 2011 Apr;95(5):1589-94. Epub 2010 Aug 24. |
|
Nik-Zainal S, Strick R, Storer M et al: High incidence of recurrent copy number variants in patients with isolated andsyndromic Mullerian aplasia. J Med Genet. 2011 Mar;48(3):197-204. Epub 2011 Jan 28. |
|
Sundaram UT, McDonald-McGinn DM, Huff D et al: Primary amenorrhea and absent uterus in the 22q11.2 deletion syndrome. Am J Med Genet A. 2007 Sep 1;143A(17):2016-8. |
|
Gervasini C, Grati FR, Lalatta F et al: SHOX duplications found in some cases with type I Mayer-Rokitansky-Kuster-Hausersyndrome. Genet Med. 2010 Oct;12(10):634-40. |
|
Belin V, Cusin V, Viot G et al: SHOX mutations in dyschondrosteosis (Leri-Weill syndrome). Nat Genet. 1998 May;19(1):67-9. |
|
Shears DJ, Vassal HJ, Goodman FR et al: Mutation and deletion of the pseudoautosomal gene SHOX cause Leri-Weilldyschondrosteosis. Nat Genet. 1998 May;19(1):70-3. |
|
Sandbacka M, Halttunen M, Jokimaa V et al: Evaluation of SHOX copy number variations in patients with Mullerian aplasia. Orphanet J Rare Dis. 2011 Aug 2;6:53. |
|
Biason-Lauber A, Leiberman E, Zachmann M: A single amino acid substitution in the putative redox partner-binding site of J Clin Endocrinol Metab. 1997 Nov;82(11):3807-12. |
|
Vainio S, Heikkila M, Kispert A et al: Female development in mammals is regulated by Wnt-4 signalling. Nature. 1999 Feb 4;397(6718):405-9. |
|
Kucheria K, Mohapatra I, Ammini AC et al: Clinical and DNA studies on 46, XY females with gonadal dysgenesis. A report ofsix cases. J Reprod Med. 1996 Apr;41(4):263-6. |
|
Resendes BL, Sohn SH, Stelling JR et al: Role for anti-Mullerian hormone in congenital absence of the uterus and vagina. Am J Med Genet. 2001 Jan 15;98(2):129-36. |
|
van Lingen BL, Reindollar RH, Davis AJ, et al.: Physical mapping of a chromosomal translocation breakpoint in a patient with congenital absence of the uterus and vagina. J Soc Gyn Invest 5:F420, 1998c |
|
Amesse L, Yen FF, Weisskopf B et al: Vaginal uterine agenesis associated with amastia in a phenotypic female with a denovo 46,XX,t(8;13)(q22.1;q32.1) translocation. Clin Genet. 1999 Jun;55(6):493-5. |
|
Park IJ, Jones HW Jr. A new syndrome in two unrelated females: Klippel-Feil deformity, conductive deafness, and absent vagina. Birth Defects 7: 311, 1971 |
|
Nager GT, Chen SCA, Hussels IE: A new syndrome in two unrelated females: Klippel-Feil deformity, conductive deafness, and absent vagina: Case II. Birth Defects 7: 312, 1971 |
|
Hillemand B, Bonneau JC, Joly JP: Conjonction d'un syndrome de Turner, d'un syndrome de Rokitansky-Kuster-Hauser atypique et d'un syndrome de Klippel-Feil. Ann Med Interne 124: 423, 1973 |
|
Baird PA, Lowry RB: Absent vagina and the Klippel-Feil anomaly. Am J Obstet Gynecol 118: 290, 1974 |
|
Mecklenburg RS, Krueger PM: Extensive genitourinary anomalies associated with Klippel-Feil syndrome. Am J Dis Child 128: 92, 1974 |
|
Letterie GS, Vauss N: Müllerian tract abnormalities and associated auditory defects. J Reprod Med 36: 765, 1991 |
|
Nykiforuk NE: Uterus didelphys. Can Med Assoc J 38: 175, 1938 |
|
Tyler GT: Didelphys in sisters. Am J Surg 45: 337, 1939 |
|
Polishuk WZ, Ron MA: Familial bicornuate and double uterus. Am J Obstet Gynecol 119: 982, 1974 |
|
Verp MS, Simpson JL, Elias S et al: Heritable aspects of uterine anomalies: I. Three familial aggregates with müllerian fusion anomalies. Fertil Steril 40: 80, 1983 |
|
Way S: The influence of minor degrees of failure of fusion of the müllerian ducts on pregnancy and labor. J Obstet Gynaecol Br Emp 52: 325, 1945 |
|
Holmes JA: Congenital abnormalities of the uterus and pregnancy. Br Med J 1: 1144, 1956 |
|
Stevenson AC, Dudgeon MY, McCluire HI: Observations on the results of pregnancies in women resident in Belfast: II. Abortions, hydatidiform males, and ectopic pregnancies. Ann Hum Genet 23: 395, 1959 |
|
Hay D: Uterus and unicollis and its relationship to pregnancy. J Obstet Gynaecol Br Emp 68: 361, 1961 |
|
Ergun A, Pabuccu R, Atay V et al: Three sisters with septate uteri: Another reference to bidirectional theory. Hum Reprod 12: 140, 1997 |
|
Elias S, Simpson JL, Carson SA et al: Genetic studies in incomplete müllerian fusion. Obstet Gynecol 63: 276, 1984 |
|
Stern AM, Gall JC Jr, Perry BL et al: The hand-foot-uterus syndrome: A new hereditary disorder characterized by hand and foot dysplasia, dermatoglyphic abnormalities, and partial duplication of the female genital tract. J Pediatr 77: 109, 1970 |
|
Poznanski AK, Kuhns LR, Lapides J et al: A new family with the hand-foot-genital syndrome: A wider spectrum of the hand-foot-uterus syndrome. Birth Defects 11: 127, 1975 |
|
Giedion A, Prader A: Hand-foot-uterus (HFU) syndrome with hypospadias: The hand-foot-genital (HFG) syndrome. Pediatr Radiol 4: 96, 1976 |
|
Donnenfield AE, Schrager DS, Corson SL: Update on a family with hand-foot-genital syndrome: Hypospadias and urinary tract abnormalities in two boys from the fourth generation. Am J Med Genet 44: 482, 1992 |
|
Fryns JP, Vogels D, Decock P et al: The hand-foot-genital syndrome: On the variable expression in affected males. Clin Genet 43: 232, 1993 |
|
Verp MS: Urinary tract abnormalities in hand-foot-genital syndrome. Am J Med Genet 32: 555, 1989 |
|
Mortlock DP, Innis JW: Mutation of HOXA13 in hand-foot-genital syndrome. Nat Genet 15: 179, 1997 |
|
Mortlock DP, Post LC, Innis JW: The molecular basis of hypodactyly (Hd): A deletion in HOXA13 leads to arrest of digital arch formation. Nat Genet 13: 284, 1996 |
|
Goodman FR, Bacchelli C, Brady AF et al: Novel HOXA13 mutations and the phenotypic spectrum of hand-foot-genital syndrome. Am J Hum Genet. 2000 Jul;67(1):197-202. |
|
McIlroy DM, Ward IV: Three cases of imperforate hymen occurring in one family. Proc R Soc Med 23: 633, 1930 |
|
Usta IM, Awwad JT, Usta JA et al: Imperforate hymen: report of an unusual familial occurrence. Obstet Gynecol. 1993 Oct;82(4 Pt 2 Suppl):655-6. |
|
Stelling JR, Gray MR, Davis AJ et al: Dominant transmission of imperforate hymen. Fertil Steril. 2000 Dec;74(6):1241-4. |
|
Georgy JM, Viechnicki MB: Absence of an ovary and uterine tube. Obstet Gynecol 44: 441, 1974 |
|
Simpson JL, Horwith M, Morillo-Cucci G et al: Bilateral anorchia: Discordance in monozygotic twins. Birth Defects 7: 196, 1971 |
|
Patsavoudi E, Magre S, Castanier M et al: Dissociation between testicular morphogenesis and functional differentiation ofLeydig cells. J Endocrinol. 1985 May;105(2):235-8. |
|
Jones MH, Furlong RA, Burkin H et al: The Drosophila developmental gene fat facets has a human homologue in Xp11.4 Hum Mol Genet. 1996 Nov;5(11):1695-701. |
|
Josso N: Biology and genetics of anti-mullerian hormone. Adv Exp Med Biol. 2011;707:83-5. |
|
Josso N, Belville C, di Clemente N et al: AMH and AMH receptor defects in persistent Mullerian duct syndrome. Hum Reprod Update. 2005 Jul-Aug;11(4):351-6. Epub 2005 May 5. |
|
Imbeaud S, Carre-Eusebe D, Rey R et al: Molecular genetics of the persistent mullerian duct syndrome: a study of 19families. Hum Mol Genet. 1994 Jan;3(1):125-31. |
|
Belville C, Josso N, Picard JY: Persistence of Mullerian derivatives in males. Am J Med Genet. 1999 Dec 29;89(4):218-23. |
|
Belville C, Josso N, Picard JY: Persistence of Mullerian derivatives in males. Am J Med Genet. 1999 Dec 29;89(4):218-23. |
|
Behringer RR, Cate RL, Froelick GJ et al: Abnormal sexual development in transgenic mice chronically expressing mullerianinhibiting substance. Nature. 1990 May 10;345(6271):167-70. |
|
Lee MM, Donahoe PK, Hasegawa T et al: Mullerian inhibiting substance in humans: normal levels from infancy toadulthood. J Clin Endocrinol Metab. 1996 Feb;81(2):571-6. |
|
Rey RA, Lhomme C, Marcillac I et al: Antimullerian hormone as a serum marker of granulosa cell tumorsof the ovary: Am J Obstet Gynecol. 1996 Mar;174(3):958-65. |
|
Chillon M, Casals T, Mercier B et al: Mutations in the cystic fibrosis gene in patients with congenital absence of thevas deferens. N Engl J Med. 1995 Jun 1;332(22):1475-80. |
|
McCallum T, Milunsky J, Munarriz R et al: Unilateral renal agenesis associated with congenital bilateral absence of the vasdeferens: phenotypic findings and genetic considerations. Hum Reprod. 2001 Feb;16(2):282-8. |