Metabolic and Endocrinologic Effects of Steroidal Contraception
Authors
INTRODUCTION
Each day, 12 million healthy women across the United States and 100 million worldwide take an oral medication to prevent unwanted pregnancy.1, 2 The ancillary health benefits associated with oral contraception are significant and include a reduction in the incidence of ovarian and endometrial cancers, diminished severity of benign breast disease, and protection from some forms of pelvic inflammatory disease. Clinical experience with contraceptive steroids spans almost 50 years. Although these synthetic agents were first commercially marketed in 1960, it was not until the late 1970s that a dose-response relationship between steroid dose and the incidence and severity of associated complications was appreciated. In the years that have followed, the impact of steroid contraception on metabolic and endocrine parameters has been the subject of intense investigation and has resulted in the gradual lowering of steroid dose, as well as the search for novel progestogens to lessen adverse metabolic impact.
High-dose estrogen formulations of oral contraceptives (OCs) were linked initially with an increased incidence of thromboembolic disease, which eventually led to an understanding of the profound effect estrogen has on the coagulation cascade. As a result, daily estrogen doses have been reduced nearly 80% – from 150 μg in the initial OC formulation to 20 μg/day in the lowest dose formulation. The risk of venous thromboembolism has dramatically declined with decreasing doses of estrogen,3 and it appears that the risk of acceleration of arterial disease is no longer present in healthy users of low-dose OCs.4 However, oral progestogens are known to produce changes in lipoprotein metabolism that mirror those lipoprotein profiles that place an individual at high risk for cardiovascular disease. Progestogens are also thought to be primarily responsible for the carbohydrate intolerance and insulin resistance noted with OC use; and as discussed later, high circulating insulin levels are thought by some to be atherogenic.
The purpose of the present chapter is to discuss some of the current studies pertaining to metabolic and endocrinologic effects of the newer progestogens in comparison with the old generation of progestogens. A summary of structure-function relationships and biologic effects of both the old and new generation of progestogens precedes this discussion.
NOVEL PROGESTOGENS
The pharmacology, physiology, and mechanisms of action of synthetic steroids in fertility regulation are discussed in detail elsewhere in this text. Three novel progestogens have undergone clinical trials, in part in an effort to diminish the putative androgen receptor-mediated effect of progestogens on carbohydrate and lipid metabolism. All synthetic progestogens are structurally related to either 17α-hydroxyprogesterone or 19-nortestosterone. The latter group of progestogens is subdivided further, on the basis of nomenclature of the current progestogens, to estranes and gonanes. Included in the discussion of novel progestogens is drospirenone, a synthetic progestin that is an analog of spironolactone and has biochemical and pharmacologic profiles similar to endogenous progesterone. A brief characterization of contraceptive progestogens follows.
Pregnanes
Acetylation of the 17-hydroxyl group of 17-hydroxyprogesterone gives oral potency, and substitution at the 6 position to inhibit metabolism produces a group of progestational steroids with potent oral as well as parenteral activity (Fig. 1). Derivatives with substitutions at the 17 and 6 positions include medroxyprogesterone acetate (Provera), chlormadinone acetate, and megestrol acetate. Cyproterone acetate, another compound in this class, is not only a potent progestogen but is also an extremely active antiandrogen. It is available outside the United States for treatment of hyperandrogenism (see Fig. 1).
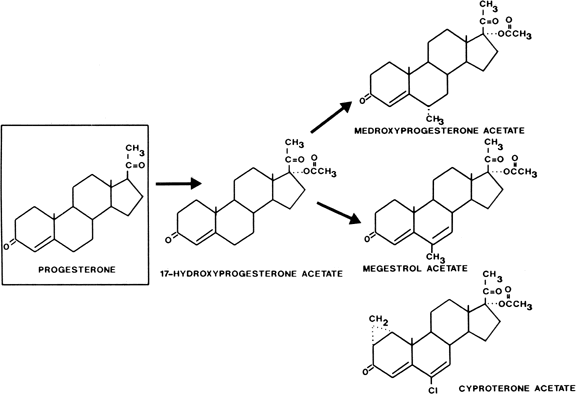{kind=link}
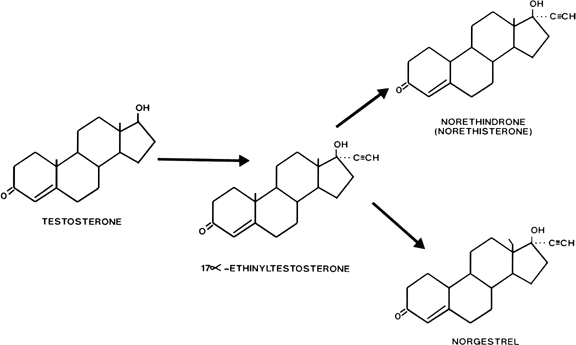
Estranes
Estranes are chemically characterized by the absence of a methyl group between rings A and B, and the presence of a methyl group at carbon 13 and an ethinyl group in the 17α position. The first orally active estrane to be synthesized was norethindrone (European name, norethisterone) (Fig. 2). Norethynodrel differs from norethindrone by having a double bond between carbons 5 and 10 instead of between carbons 4 and 5. Acetylation of the 17-hydroxyl group of norethindrone gives rise to norethindrone acetate, and acetylation at position 3 of the latter compound produces ethynodiol diacetate. Elimination of the oxygen at position 3 of norethindrone gives rise to lynestrenol. Norethynodrel, norethindrone acetate, ethynodiol diacetate, and lynestrenol are metabolized to norethindrone to produce their biologic effects (Fig. 3).
{kind=link}
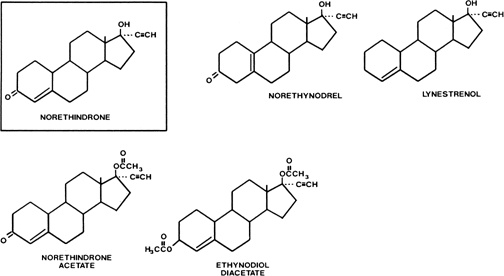{kind=link}
Gonanes
Gonanes are characterized chemically by the absence of a methyl group between rings A and B, and the presence of an ethyl group at carbon 13 and an ethinyl group at carbon 17 on the basic steroid nucleus. Substitution of an ethyl group for the methyl group at carbon 13 of norethindrone produces norgestrel, one of the most potent oral progestogens (see Fig. 2). Because this modification supposedly confers significant androgenicity to the compound, a new generation of orally active progestogens has been derived from this structure (Fig. 4). Addition of a methylene group at carbon 11 and elimination of the oxygen at carbon 3 produce desogestrel. Norgestimate results from the formation of an oxime at position 3 and an acetate at position 17. Gestodene differs from levonorgestrel only by the formation of a double bond between carbons 14 and 15.
{kind=link}
Drospirenone
Drospirenone is a novel progestin that is derived from its parent compound spirolactone, and is an analog of spironolactone. Unlike the estranes and gonanes, it contains a methyl group at carbon 10 and lacks an ethinyl group at carbon 17. Similar to spironolactone, drospirenone has both antimineralocorticoid and antiandrogenic properties. Its antimineralocorticoid action results from counteraction of the renin-angiotensin-aldosterone system, causing moderately increased sodium and water excretion. The antiandrogenic action of drospirenone is a consequence of its ability to block the binding of the two potent androgens, testosterone and dihydrotestosterone to the androgen receptor. Combined with ethinyl estradiol in oral contraceptive formulations, drospirenone-containing contraceptives have similar efficacy and safety profiles to other low-dose oral contraceptives, but appear to offer improved tolerability with regard to weight gain, mood changes, acne, and treatment of the premenstrual dysphoric disorder, due to the properties of drospirenone described above.5
SPECTRUM OF BIOLOGIC EFFECTS
The use of progestogens structurally related to progesterone in OCs was abandoned when it was discovered that chlormadinone acetate was associated with the development of breast tumors in beagle dogs.6 Progestogens currently used in OCs in the United States are the estranes and gonanes, which possess a broad range of biologic activity. Synthetic progestogens have, in various degrees, estrogenic, antiestrogenic, androgenic, and antiandrogenic effects.
Progestogenic efficacy is frequently measured with respect to ovulation inhibition, binding affinity to the progesterone receptor, induction of secretory endometrium, and pregnancy maintenance. After reviewing the published literature, Dorflinger7 charted the overall progestogen potency of various OC formulations available in the United States. She reported the progestogenic potency of the estrane preparations to be essentially equal and 10–20 times less potent than levonorgestrel. However, there are approximately 27 different measurable effects of synthetic progestogens about which a bioassay can be constructed, and depending on which specific biologic parameter is tested, the relative potency of the progestogens varies greatly.
POTENCY TESTS
Clinical effects of progestogens are sometimes based on data obtained from in vitro receptor binding assays and bioassays. However, such tests are often performed on animal models, which may not accurately reflect the clinical effects observed in women. In one widely cited report, Phillips and coworkers8 estimated progestational activity of the various progestogens by measuring binding to the rabbit uterine progesterone receptor. A similar study, however, performed by Juchem and coworkers9 using the human progesterone receptor, shows markedly different results. In both studies, levonorgestrel and 3-ketodesogestrel bound with high affinity to the receptor. However, norgestimate and two of its metabolites (levonorgestrel 3-oxime (norelgestromin) and levonorgestrel-17-acetate) bound with much higher affinity to the rabbit receptor than to the human receptor, indicating that the rabbit model overestimates the clinical effect in humans for these compounds.
Determinations of androgenic activity of progestogens have been equally problematic. Conclusions about androgenic effects of various progestogens are often based on data from animal studies or on serum measurements of sex hormone-binding globulin (SHBG) or free testosterone. The study of progestogen binding to androgen receptors in the rat prostate by Phillips and associates8 has been cited as showing that levonorgestrel binds more avidly to androgen receptors than do the newer progestogens. Extrapolating these data to the human female is controversial, however, because women do not have an androgen-responsive tissue comparable to the prostate. Similarly, measurements of SHBG are used to hypothesize decreased androgenic effects of the new progestogens. However, it is well recognized that the estrogenic component of combined OCs increases circulating SHBG levels, thereby lowering the levels of free testosterone. Additionally, our recent data (unpublished) show a substantial suppression of ovarian testosterone production after administration of levonorgestrel combined with ethinylestradiol (EE2) to women resulting in a further lowering of the free testosterone concentration.
A more meaningful way to assess androgenicity of OCs is to determine their effect on female tissues that respond to androgens (e.g., the skin). A key element of hair growth in skin is the pilosebaceous unit. Cell nuclei in this unit contain the pivotal enzyme, 5α-reductase, which transforms androstenedione or testosterone to dihydrotestosterone (DHT). DHT is the most potent endogenous androgen and the hormone responsible for clinical manifestations of hirsutism, acne, or alopecia. In an in vitro study by Cassidenti and associates,10 the formation of DHT from testosterone in genital skin was inhibited by levonorgestrel and norethindrone. The results showed approximately 48% inhibition of 5α-reductase activity by levonorgestrel and 59% inhibition by norethindrone, suggesting that these progestogens are potent antiandrogens with respect to their action on the pilosebaceous unit.
One of the most important measurable effects of progestogens is ovulation inhibition. Progestogen doses required for ovulation inhibition have been determined for various combination OCs.11 The data show that high doses are required for norethindrone (400 mg) and norgestimate (200 mg), and relatively low doses for gestodene (30 mg), levonorgestrel (60 mg), and desogestrel (60 mg).
PHARMACOKINETICS
Serum levels of progestogens can be measured by radioimmunoassay and used to determine the half-life and bioavailability of a progestogen. In general, the half-life of levonorgestrel is approximately twice that of norethindrone, whereas the half-lives of 3-ketodesogestrel and gestodene are similar to the half-life of levonorgestrel.12
Bioavailability is the extent to which a drug enters the circulation after its first pass through the liver. Studies show that the bioavailability of norethindrone is approximately 64%, and the other estranes, which include norethindrone acetate, ethynodiol diacetate, norethynodrel, and lynestrenol, are all converted to norethindrone, which serves as the active progestational metabolite for these prodrugs.12 As for the gonanes, both levonorgestrel and gestodene have bioavailabilities that are greater than 90%, whereas the bioavailability of desogestrel, which undergoes hepatic transformation to its progestationally active metabolite, namely, 3-ketodesogestrel (etonogestrel), is approximately 62%.12 The bioavailability of norgestimate, however, cannot be determined because it is metabolized extensively to two different metabolites (norelgestromin and levonorgestrel).13 However, it has been shown that approximately 25% of levonorgestrel is derived from orally administered norgestimate.14 Although norelgestromin is found in relatively high concentrations in the circulation, its binding to the human progesterone receptor is very low. A third norgestimate metabolite, levonorgestrel-17-acetate, has substantial affinity for the human progesterone receptor, but its concentration in blood is barely detectable (unpublished data).
PROGESTOGEN-ESTROGEN INTERACTION
It is important to realize that biologic effects of progestogens in combined OCs are a consequence of their synergistic relationship with the estrogenic component of the OC. Several years ago, controversy arose from a published report by Jung-Hoffman and Kuhl15 on the influence of gestodene and desogestrel on the metabolism of EE2. Their report concluded that gestodene, but not desogestrel, impedes the metabolism of EE2 in women after administration of OCs containing EE2 in combination with gestodene or desogestrel. Closer examination of the conclusions drawn by Jung-Hoffman and Kuhl showed that on the basis of in vitro data,16 17-ethinylated progestogens at high concentrations can inhibit EE2 hydroxylation in hepatic microsomes. However, in vivo, there is no direct evidence to support the view that EE2 metabolism is inhibited or that one progestogen is more inhibitory than another.13
CARBOHYDRATE METABOLISM
Both estrogens and progestogens modify carbohydrate metabolism and, as with other metabolic parameters, the net effect on glucose use is the result of the balance of these two components in a given formulation. In light of the pharmacologic doses of both estrogen and progestogen in the initial OC preparations, in retrospect, it is not surprising that glucose intolerance surfaced as a concern as early as 1963.17
Small amounts of estrogen improve carbohydrate metabolism18 and may even improve hyperglycemia in noninsulin-dependent diabetes.19 The mechanism underlying this improvement may be the alteration of insulin receptor binding20 or possibly the inhibition of insulin degradation.21
In contrast, progesterone in high doses and synthetic progestogens in moderate to low doses cause impairment of glucose use and insulin resistance.22 Progesterone-induced changes in carbohydrate metabolism are typified by the emergence of insulin resistance in the third trimester of pregnancy, in which there is an exaggerated insulin response to a glucose load, with subsequent blunted response to hypoglycemia.
Epidemiologic studies show a strong association between elevations of both glucose and insulin and an increased risk of coronary artery disease, and women with impaired glucose tolerance appear to be at even greater risk of having heart disease develop than diabetic men.23 High circulating levels of insulin have been implicated in atherogenesis. Insulin is known to stimulate cell proliferation and lipid synthesis in arterial smooth muscle24 and has been shown to promote atheroma formation in vivo.25
The classic method of measuring glucose tolerance is performance of an oral glucose tolerance test, and insulin resistance is measured with the euglycemic hyperinsulinemic clamp. The greatest impairment of glucose use was noted during therapy with the initial high-estrogen/high-progestogen OC formulations. Patients taking these combinations typically showed a low fasting plasma glucose level, decreased glucose tolerance, especially in the first hour after a glucose challenge, and diminished early insulin response with subsequent enhanced insulin secretion.26 At these higher estrogen doses (50–150 μg/day), estrogen is thought to have a direct effect on the liver and pancreas, resulting in an overall decreased portal secretion of insulin and glucagon with an increased insulin:glucagon ratio.27
Some deterioration of glucose use is evident with even moderate doses of progestogens, such as those found in the monophasic low (estrogen)-dose pills.28, 29 Certain researchers believe that the degree of deterioration of glucose homeostasis varies according to the compound's intrinsic androgenicity and that it increases in series: pregnanes, estranes, and gonanes.30 Others think that in terms of inducing adverse carbohydrate effect, there is no inherent difference between biochemical classes of progestogens, but rather that the issue involves using equipotent doses.31
In a series of experiments, Spellacy and colleagues28, 29 showed that progestogens are primarily responsible for the deterioration of insulin response, showing that the progestogen-only “mini-pill” produced insulin elevations. These experiments also showed the importance of the balance of the steroid effects on net glucose use by demonstrating that the corresponding progestogen in combination with low-dose estrogen induced fewer changes in carbohydrate metabolism than when used alone.
Progesterone and synthetic progestogens have been shown to stimulate insulin release directly from pancreatic β-cells in animal models.32 The mechanism of insulin resistance induced by progestogens is currently under debate. Although some researchers have shown decreased peripheral insulin receptor concentrations and affinity in monophasic pill users,31, 33 others have been unable to confirm this finding.34 Research to examine possible alterations in transmembrane signaling/postreceptor defects is under way.
The amount of progestogen exposure is emerging as a critical element in the induction of altered glucose use. Skouby35 compared the effect of two levonorgestrel preparations on glucose use, a monophasic OC formulation and a triphasic formulation that reduced the content of levonorgestrel by approximately 30%. In contrast to the hyperinsulinemic response to a glucose challenge noted in the patients taking the monophasic formulation, no changes in glucose use were noted with the triphasic formulation. This is consistent with several other reports showing minimal or no deterioration of glucose homeostatis with the triphasic OCs,36, 37, 38 even in women with a history of gestational diabetes.39
In a large cross-sectional study by Godsland and coworkers,40 1060 women took one of nine types of OCs for at least 3 months and 418 women took none. Seven of the contraceptive formulations contained various doses (50–100 μg) and types of progestogens (e.g., levonorgestrel, norethindrone) as either a monophasic or triphasic preparation in combination with 30–40 μg EE2. Fasting plasma glucose levels were either reduced or moderately increased, whereas fasting insulin and C-peptide levels were generally increased slightly. On the basis of available data, it appears that changes in glucose and insulin levels with low-dose OCs are slight but not clinically meaningful, with no increased risk of diabetes mellitus or coronary disease being observed in OC users.
LIPID AND LIPOPROTEIN METABOLISM
Contraceptive steroids have a diverse impact on lipid and lipoprotein metabolism. Individuals with certain genetic and dietary-related lipoprotein profiles have been shown to be at significantly increased risk of morbidity from atherosclerosis and cardiovascular disease.41, 42, 43 An important epidemiologic study of the Royal College of General Practitioners showed that with the dose of estrogen remaining constant, an increase in the progestogen dose in the early OC formulations was associated with both a dose-related decrease in high-density lipoproteins (HDLs) and an increase in the rate of total arterial disease.44
Despite the epidemiologic data, there is no direct experimental evidence that OC-induced alterations in lipid parameters lead to an increase in atherosclerosis. It has been hypothesized that any adverse alteration in lipid metabolism may accelerate the development of this disease, and therefore the search for lipid-neutral contraceptive formulations has been undertaken. A basic understanding of lipid physiology is necessary to interpret these studies.
Lipids, which are insoluble in plasma, combine with proteins to form soluble complexes called lipoproteins. Lipoproteins are spherical molecules that contain four basic components: cholesterol, triglyceride, phospholipid, and proteins known as apoproteins or apolipoproteins. Cholesterol is carried both in a free form on the lipoprotein's surface and within the core of the lipoprotein complex as cholesteryl ester. Triglyceride is a nonpolar fat carried within the core of the lipoprotein complex. Phospholipids and apoproteins are electrically charged and reside on the surface of the molecule. They provide the polarity and resultant miscibility of the complex that allows for its transport in plasma. Apoproteins play a central role in the use and metabolism of circulating lipids. Specialized apoproteins act as receptor sites on lipoproteins for the uptake of nontissue-bound cholesterol. Others are recognized by specific receptors on target cells, which bind the lipoprotein complex, making the cholesterol available to the cell for synthesis of steroids or plasma membranes, or for storage. Apoproteins also act as cofactors or inhibitors of various enzymes affecting lipoprotein use.
CARDIOVASCULAR EFFECTS
In general, estrogen tends to have an effect on the lipid profile that theoretically would offer protection from development of cardiovascular disease by producing an increase in high-density lipoproteins (HDL) and a decrease in low-density lipoproteins (LDLs). Progestogen, conversely, exhibits an opposite and adverse effect decreasing HDL and increasing LDL.
Exogenous estrogens increase plasma HDL concentrations, specifically increasing the HDL subfraction HDL, apoprotein A-I, and plasma apoprotein A-I.45 They achieve this in part by decreasing hepatic endothelial lipase activity46 and diminishing HDL catabolism. Pharmacologic doses of estrogen also decrease plasma LDL concentrations, presumably by upregulation of hepatic LDL receptors.47 Unfortunately, estrogens also induce an increase in the synthesis of very-low-density lipoproteins (VLDLs) leading to elevations in serum triglyceride levels.48
19-nortestosterone-derived progestogens oppose the effects of estrogen on lipoprotein physiology.49 These progestogens increase HDL catabolism by increasing hepatic endothelial lipase activity.50 They decrease concentrations of VLDL/triglyceride and increase LDL concentrations in the plasma. A unifying concept of these effects was proposed by Khokha and colleagues.50 By increasing hepatic lipase activity, progestogens enhance VLDL/triglyceride degradation, resulting in increased hepatic supply of cholesterol, downregulation of LDL receptors in the liver, and increased LDL release into the circulation.
Because the effects of synthetic progestogens on lipoprotein metabolism are opposite those of estrogen, both the ratio of estrogen to progestogen and the degree of estrogenicity and androgenicity of the progestogen determine the extent and direction of OC-induced alterations in lipid physiology. LDL concentrations are proportionately higher in women ingesting formulations that contain the lowest estrogen:progestogen ratio. Triglyceride and HDL levels are proportionately higher with OCs that contain higher amounts of estrogen or less antiestrogenic progestogen. To produce a lipid-neutral formulation, researchers have sought to identify the proper dose and chemical structure of a progestogen to offset the estrogenic increase in triglycerides without adversely affecting the other lipid parameters.
As the understanding of the effects of contraceptive steroids on lipid and carbohydrate metabolism has evolved, the formulations of OCs have reflected this knowledge, first by adjusting the balance of the estrogenic component to the progestogenic component and, more recently, by developing triphasic preparations that reduce the total exposure to the progestogen, as well as developing novel progestogens that show equivalent contraceptive efficacy with fewer androgenic effects.
An example of a favorable estrogen:progestogen balance is found in the lowest-dose combination estrane formulation containing EE2 (35 μg) and norethindrone (400 μg), which shows no significant increase in the levels of total cholesterol or triglycerides, with a trend toward increased levels of HDL3 without a concomitant decrease in HDL2.51 Similarly, in studies of the triphasic combination containing mean daily doses of EE2 (32 μg) and levonorgestrel (92 μg), no significant changes in the lipid and lipoprotein concentrations are noted,52 except for an increase in the level of HDL3. However, this increase in the HDL3 subfraction appears to occur at the cost of a reduction of the HDL2 subfraction which is thought to have greater cardioprotective activity.53 In contrast, formulations of desogestrel compared with levorgestrel reveal an estrogen-dominant effect on lipoprotein metabolism. When compared with triphasic levonorgestrel combinations, even higher mean daily progestogen exposures of monophasic desogestrel (150 μg) produce less antagonism of the estrogen-stimulated decreases in LDL and increases in HDL (both HDL and HDL subfractions), but they are associated with moderate increases in triglycerides.54
It should be noted that the changes in lipid values induced by OCs are typically within the normal range for women and of uncertain predictive value regarding serious clinical consequence. If the lipid hypothesis were correct, atheromatous changes in vessel structure occurring during OC use should eventually place those women at greater risk of coronary heart disease than women who have never used OCs. Although there have been some reports of increased risk of cardiovascular disease, especially among women who smoked while ingesting OCs containing high progestogen doses,55 the majority of published epidemiologic studies indicate that there is no increased risk of myocardial infarction among former users of OCs.56, 57 Further, the incidence of cardiovascular disease is not correlated with the duration of OC use.58
Experimental evidence that combination OC-induced alterations in lipid physiology lead to atherosclerosis is also lacking. In animal studies, macaque monkeys were maintained for a period of 2 years on an OC formulation containing EE2 and levonorgestrel, which reduced HDL concentrations to levels that would presumably accelerate the development of atherosclerosis.59 The animals were at the same time being fed an atherosclerogenic diet. At necropsy, despite their low HDL values, OC-treated female macaques had less-extensive atherosclerosis than did untreated control animals. Another group of monkeys, treated with levonorgestrel without estrogen, also had lowered HDL level. In this group, extent of coronary atherosclerosis was significantly greater than in control subjects.
The results of the studies on macaques, confirmed in a larger study that also examined the effects of the progestogen, ethynodiol diacetate,60 suggest that the estrogen component of OCs may actually protect the user against coronary atherosclerosis despite changes in lipid profiles such as raised serum triglyceride and LDL cholesterol levels, reduced HDL cholesterol levels, impairment of glucose tolerance, and elevated insulin levels that would otherwise appear to be contributory to cardiovascular disease. A study by Godsland and coworkers confirmed that a reduction in the dose of progestogen and a change in the type of progestogen can bring a substantial reduction in risk markers for coronary heart disease in users of low estrogen-containing OCs.40 However, the general conclusion from that study was that levonorgestrel tends to decrease HDL and increase LDL levels slightly, whereas desogestrel and norethindrone have the opposite effect. The changes, however, are minor and do not appear to be clinically meaningful with regard to the risk of subjects having cardiovascular disease develop.
The exact mechanism of the protective effect of estrogen is currently under investigation. As well as modifying the relative amount of circulating LDL, the principal carrier of cholesterol in the bloodstream, estrogen has been shown to alter its composition, increasing the content of triglycerides and phospholipids61 and resulting in a less cholesterol-rich molecule, which may reduce the uptake of cholesterol in vessel walls. Estrogen receptors have been shown to be present in vessel walls in several mammalian species. Effects of estrogen at the level of the arterial lumen include reductions in arterial smooth-muscle cell proliferation,62 decreased production and increased degradation of collagen and elastin,63 and modification of prostacyclin and thromboxane synthetase.64 This protection from cardiovascular morbidity may be negated by smoking. Milekowsky and associates64 showed that smoking reduces prostacyclin production while increasing thromboxane production, thereby leading to vasospasm of susceptible vessels.
COAGULATION EFFECTS
One of the first complications of estrogen-containing OCs to be noted was an increase in the incidence of thromboembolic disease. EE2 causes dose-dependent alterations in the hepatic production of several globulins involved in the coagulation process. The vitamin K-dependent clotting factors II, VII, IX, and X are elevated,65 and antithrombin II production is decreased.66 However, unlike the marked aberrations noted with the first OC formulations, doses of less than 50 μg are associated with little or no impact on the various clotting factors.67 It is thought that slight increases in thrombin formation are offset by increased fibrinolytic activity and that platelet activation and clotting therefore are not increased.68
Differences in risk of deep vein thrombosis (DVT) in third-generation (gestodene, desogestrel, and norgestimate with 35 μg EE2 or less) versus second-generation (levorgestrel with 35 μg EE2 or less) OC pills in studies performed in the United Kingdom and Germany showed that differences in the risk of DVT were observational, subject to bias, and likely not causally related.69 If increase in DVT with third-generation OCs is believed to be causally related, users should be so informed. If they are informed of the risk of increased DVT, they should also be advised that the same studies showed one third less risk of myocardial infarction and coronary vascular disease with third-generation versus second-generation death from OCs. In its only written communication concerning this matter, the US Food and Drug Administration (FDA) stated that it “does not recommend that women using the desogestrel containing products stop using them or change to another oral contraceptive.”70
Patients with inherited thrombophilias typically present with DVT of the legs, pulmonary embolism, or both usually before the age of 45 years. Mutation of factor Va gene (factor V Leiden) is a major cause of activated protein C (APC) resistance, occurs in 3–5% of the population, and has been found to be present in 20% of patients with DVT. APC is anticoagulatory; it cleaves factors Va and VIIa. The relative risk of DVT with APC resistance is increased eight times above the norm. The relative risk of DVT in OC users with APC resistance increases 35 times above the norm.71 Therefore, women with inherited thrombophilias should avoid using OCs, particularly if they have a personal or family history of venous thrombosis.72 In the absence of a clear family history of venous thrombosis, there is little justification to screen for prothrombotic mutations. More than half a million women would need to be screened for factor V Leiden to avoid a single death from pulmonary embolus since only 5–10% of white women are carriers, which is far less common in women of Asian or African origin, and the mortality associated with venous thrombosis in young women is low.72, 73, 74
HYPERTENSION
Approximately 5% of women using early, high-dose formulation OCs had hypertension develop,75 and a significant elevation in blood pressure is still observed in some women using formulations with the 30–35-μg EE2 dose.76 Estrogen is primarily responsible for these blood pressure elevations, but there is evidence that progestogens may play a contributory role.
Estrogens induce the hepatic production of renin substrate, angiotensinogen, with a secondary increase in angiotensin.77 The elevation of aldosterone that results causes sodium and water retention. These are dose-related effects. With current OC formulations, most women experience only minor elevations in blood pressure and remain normotensive. If severe hypertension develops, it typically resolves 3–4 months after discontinuation of treatment.
Progesterone is known to increase sodium excretion. It has high affinity for the aldosterone receptor and competes with aldosterone at the level of the renal tubule. The synthetic progestogens do not share this biologic activity.78 Except for drospirenone, synthetic progestogens, unlike progesterone, do not inhibit the aldosterone-induced retention of sodium and have the potential for a gradual positive sodium balance and increase in blood pressure.79
Although no significant elevations in blood pressure have been noted in women receiving triphasic formulations, patients with a history of hypertension or renal disease should be monitored carefully if receiving these preparations.
WEIGHT GAIN
Studies of weight gain in women who took formulations of triphasic levonorgestrel and triphasic norgestimate daily for 6 months showed no differences between baseline and treatment values or between different progestogens.80, 81 A product using a derivative of spironolactone, an antimineralocorticoid progestogen, drospirenone, actually reported a slight decrease in weight and blood pressure in users.82
ENDOCRINOLOGIC EFFECTS
Hypothalamus-pituitary
Although contraceptive steroids interfere with the reproductive process at several levels, the primary site of action is at the level of the hypothalamus and pituitary to suppress the midcycle luteinizing hormone (LH) surge, thereby preventing ovulation. Basal secretion of LH is not suppressed, even with preparations of synthetic estrogens in a 100-μg/day dose.83 With combinations containing 50 μg of EE2 or less, irregular LH peaks can be observed throughout the treatment cycle.84
There is some evidence that pituitary reserve of gonadotropins is depleted by OC steroids. This was shown by a sequential pituitary stimulation test (SST) consisting of an infusion of thyrotropin-releasing hormone (TRH) followed by gonadotropin-releasing hormone (GnRH). The pituitary response to stimulation was significantly depressed in OC users compared with control subjects, even after GnRH priming, after ingestion of OC formulations containing both high- and low-dose synthetic estrogens.85 This effect appears to be dose related in that patients receiving less than 50 μg of estrogen had a lesser LH suppression after a bolus of GnRH.86 Responses of both growth hormone and thyroid-stimulating hormone (TSH) to the SST were unaffected by ingestion of contraceptive steroids, whereas the prolactin response was significantly elevated after treatment with either high- or low-dose estrogen therapy.
Estrogen and the pituitary lactotroph
Coincident with the advent of widespread OC use, laboratory techniques for the measurement of prolactin and radiographic technologies for the detection of pituitary lesions improved dramatically, and an association between the use of contraceptive steroids and the development of prolactin-secreting pituitary adenomas arose.
Several observations fueled speculation that the estrogen contained in OCs caused pituitary adenomas in women. There is in vitro evidence of a specific receptor of estrogen on the lactotroph that regulates the transcription of the prolactin gene.87 Endogenous estrogen stimulates prolactin secretion, causing hyperprolactinemia and lactotroph hyperplasia throughout human gestation.88 In addition, pharmacologic doses of exogenous estrogen were shown in animals studies to induce pituitary adenomas in rodents.89
However, the Pituitary Adenoma Study Group90 reporting the data from a large multicenter case-control study found no causal relationship between OC use and the risk of subjects having a prolactin-secreting adenoma develop, consistent with the findings of two large prospective studies conducted by the Royal College of General Practitioners and the Oxford Family Planning Association reported earlier.91 As the authors discuss, the data collected were not designed to address the issue of whether estrogen may exacerbate a pre-existing adenoma. A Swedish group studied 70 women with prolactinomas and compared those who had previously used OCs (61%) with those who had not.92 Patients who had used OCs had a shorter duration of symptoms, lower serum prolactin levels, and less-pronounced enlargement of the sella turcica, suggesting that the use of OCs either did not promote the growth of the pituitary adenoma or may have led to earlier detection of the lesion.
It has been reported in a number of studies that basal levels of prolactin are slightly elevated in healthy women taking OCs.93 Mishell and colleagues85 performed TRH stimulation tests on a group of women taking various doses of contraceptive estrogens and found the initial prolactin response to TRH, as well as the maximal prolactin response, to be significantly greater in subjects using high or low doses of estrogen formulations.
Episodic hyperprolactinemia, probably stress-related, is not uncommon in healthy women,94, 95 and the diagnostic precision of finding elevated prolactin levels associated with an organic lesion is improved by frequent sampling.96 Luciano and colleagues97 reported that the incidence of hyperprolactinemia as determined by multiple blood sampling was significantly greater in a group of OC users than in a group of women who used barrier methods of contraception (12% versus 5%), but that the hyperprolactinemia was often transient and resolved spontaneously in approximately 50% of both OC users and control subjects. They also identified a subset of patients who were more sensitive to the lactogenic effects of exogenous estrogens and who therefore might be at greater risk of having hyperprolactinemia develop. Consistent with other reports, neither the duration of pill use nor the dose of estrogen in the pill had any effect on the development of hyperprolactinemia.
Although the Pituitary Adenoma Study Group90 reported that the relative risk of subjects having a prolactinoma develop was not increased in OC users with a history of menstrual disorders. Luciano and colleagues97 noted that the patients in their study in whom hyperprolactinemia developed on OCs had a significantly higher prevalence of menstrual dysfunction in their histories. Regardless of whether they take OCs, women who have irregular menstrual periods are more likely to have hyperprolactinemia and are more likely to have secondary amenorrhea develop. Therefore, each individual case should be investigated before beginning OC therapy.
Return of fertility
In a review by MacLeod,98 it was found that the incidence of postpill amenorrhea and that of secondary amenorrhea unrelated to OC use were similar (0.2–2.2% and 0.1–0.8%, respectively). The rate of return of fertility after the discontinuation of OCs is lower than for women who have used barrier methods for 2–3 years, but the percentage of women who eventually conceive after ceasing either form of contraception becomes the same.99 Neither the rate of spontaneous abortion100 nor the incidence of chromosomal abnormalities101 in abortuses is increased in women who conceive in the first or subsequent months after discontinuing OCs. More important, in contrast to earlier published reports, a large cohort study reported that ingestion of OC steroids in the first few months of pregnancy does not significantly increase the risk of congenital malformations among the offspring of users overall or among those of nonsmoking users.102
Thyroid
Thyroid hormone circulates as both thyroxine (T4) and triiodothyronine (T3). Most of the circulating thyroid hormone is bound to thyroid-binding globulin, whereas less than 0.1% circulates in the unbound or free form. It is the unbound hormone that is thought to have a biologic effect. As we note in the discussion of other metabolic parameters, ingestion of synthetic estrogens stimulates hepatic production of globular proteins, including thyroid-binding protein. Therefore, total thyroxine levels are elevated with OC use, reflecting an increased bound fraction. However, levels of free thyroxine are unaffected by estrogen ingestion and, together with measurement of TSH, provide an accurate assessment of a patient's thyroid state.
Adrenals
Similar to thyroid-binding globulin, estrogen increases hepatic production of corticosteroid-binding globulin (CBG). As early as 1963, women taking OCs were noted to have significantly elevated levels of total plasma cortisol.103 It was later reported that levels of both free and bound cortisol are elevated by combination OCs.104 Both components of combination pills are involved in this increase in free cortisol.104 Estrogen reduces the ability of the liver to metabolize cortisol and progesterone, and certain progestogens can displace cortisol from CBG.105
The secretion of adrenocorticotropic hormone (ACTH) is diminished during OC therapy, possibly by negative feedback of unbound cortisol.106 Dehydroepiandrosterone sulfate (DHEAS) levels are also decreased,107 depending on the type of progestogen used. Administration of estrogen alone results in an increase in DHEAS,108 whereas OCs containing norethindrone and levonorgestrel show a suppressive effect on adrenal androgen secretion.109 Despite this putative suppression of adrenal function, the adrenal glands respond normally to ACTH in women taking OCs, and the pituitary-adrenal response to stress appears unaffected.
Skin
The triphasic EE2/norgestimate combination OC pill is the first and only OC pill indicated for the treatment of moderate acne vulgaris in menarchial women unresponsive to topical anti-acne medication. This was the first time that the FDA stated that an OC can be indicated for noncontraceptive use.110 However, more recent studies show that low-dose OCs improve acne,109 and it appears that one OC is not any better than another in improving this clinical manifestation.
The mechanism by which OCs improve acne and hirsutism can be explained by considering androgen effects at the level of the pilosebaceous unit in skin. Within these cells, androstenedione and testosterone are converted to DHT via the pivotal enzyme, 5α-reductase.111 The latter hormone is markedly more potent than testosterone. OCs suppress bioavailable testosterone, which is the non-SHBG-bound fraction of total testosterone, thereby reducing the availability of precursor for 5α-reductase.109 Studies also show that in skin of hirsute women, there is an increased conversion of testosterone to DHT compared with nonhirsute women, suggesting that increased activity of 5α-reductase underlies hirsutism more so than elevated levels of testosterone.111 Lobo and colleagues have shown in vitro that levonorgestrel and norethindrone inhibit 5α-reductase activity by 50–60%, thereby suggesting a mechanism by which progestogens may achieve their observed improvements in acne and hair growth.10 Furthermore, it throws into question the concept that certain progestogens are inherently androgenic.
REFERENCES
Forrest JD, Fordyce RR: U.S. women's contraceptive attitudes and practice: How have they changed in the 1980's. Fam Plann Perspect 20: 112, 1988 |
|
Trussell, James (2007). "Contraceptive Efficacy". in Hatcher, Robert A., et al. Contraceptive Technology (19th rev. ed.). New York: Ardent Media. ISBN 0-9664902-0-7. |
|
Meade TW, Greenberg G, Thompson SG: Progestogens and cardiovascular reactions associated with oral contraceptives and a comparison of the safety of 50- and 30-μg oestrogen preparations. Br Med J 280: 1157, 1980 |
|
Mishell DR Jr: Use of oral contraceptives in women of older reproductive age. Am J Obstet Gynecol 158: 1652, 1988 |
|
Muhn P, Krattenmacher R, Beier S, Elger W, Schillinger E. Contraception. 1995 Feb;51(2):99-110. |
|
Daniel GR: Chlormadinone contraceptive withdrawn. Br Med J 1: 303, 1970 |
|
Dorflinger LJ: Relative potency of progestins used in oral contraceptives. Contraception 31: 537, 1985 |
|
Phillips A, Demarest K, Kahn DW et al: Progestational and androgenic receptor binding affinities and in vivo activities of norgestimate and other progestins. Contraception 41: 339, 1990 |
|
Juchem M, Pollow K, Elger W et al: Receptor binding of norgestimate- a new orally active synthetic progestational compound. Contraception 47: 225, 1994 |
|
Cassidenti DL, Paulson RJ, Serafini P et al: Effects of sex steroids on skin 5α-reductase activity in vitro. Obstet Gynecol 78: 103, 1991 |
|
Teichman AT: Levonorgestrel. Stuart-Georg Thieme Verlag, 43, 1990 |
|
Stanczyk FZ: Structure-function relationships, metabolism pharmacokinetics and potency of progestens. Drugs of Today 32: 1, 1996 |
|
Stanczyk FZ: Pharmacokinetics of the new progestogens and influence of gestodine and desogestrel on ethinglestradiol metabolism Contraception 55:273, 1997 |
|
Kuhnz W, Blode H, Mahler M: Sytematic availability of levonorgestrel after single oral administration of a norgestimate containing oral contraceptive to 12 women. Contraception 47: 283, 1993 |
|
Jung-Hoffman C, Kuhl H: Interaction with the pharmacokinetics of ethinylestrodiol and progestogens contained in oral contraceptives. Contraception 40: 299, 1989 |
|
Guengerick FP: Inhibition of oral contraceptive steroid-metabolizing enzymes by steroids and drugs. Am J Obstet Gynecol 21: 159, 1990 |
|
Waine H, Frieden EW, Caplan HI et al: Metabolic effects of Enovid in rheumatoid patients (abstr). Arthritis Rheum 6: 796, 1963 |
|
Spellacy WN: Menopause estrogen treatment and carbohydrate metabolism. In Mishell DR (ed): Menopause-Physiology and Pharmacology, p 253. Chicago Year Book Medical Publishers, 1987 |
|
Kalkhoff RK: Effects of oral contraceptive agents on carbohydrate metabolism. J Steroid Biochem 6: 949, 1975 |
|
Ballejo G, Saleem TH, Khan-Dawood FS et al: The effect of sex steroids on insulin binding by target tissues in the rat. Contraception 28: 413, 1983 |
|
Tsibris JCM, Ballejo G, Kramer NC et al: Estrogens inhibit the degradation of insulin by human placental preparations in the presence of glutathione. Biochem Biophys Res Commun, 1988 |
|
Wynn V: Effects of progesterone and progestins on carbohydrate metabolism. In Bardin CW, Milgrom E, Mauvis-Jarvis P (eds): Progesterone and Progestins, pp 395–410. New York, Raven Press, 1983 |
|
Jarrett RJ: The epidemiology of coronary heart disease and related factors in the context of diabetes mellitus and impaired glucose tolerance. In Jarret RJ (ed): Metabolic Aspects of Cardiovascular Disease, pp 1–23. New York, Elsevier Biomedical Press, 1984 |
|
Goldsand I, Crook D, Stevenson J et al: Estrogen/progestin combinations and carbohydrate metabolism: Long-term risk and methods of assessment. In Lobo RA, Whitehead MI (eds): Consensus Development Conference on Progestogens. Int Proc J 1: 74, 1989 |
|
Stout RW: Insulin and Atheroma-an update. Lancet 1: 1077, 1987 |
|
Gaspard UJ: Metabolic effects of oral contraceptives. Am J Obstet Gynecol 157: 1029, 1987 |
|
Spellacy WN, Buhi WC, Birk SA: Effects of norethindrone on carbohydrate and lipid metabolism. Obstet Gynecol 46: 560, 1975 |
|
Spellacy WN, Buhi WC, Birk SA: Carbohydrate and lipid metabolic studies before and after one year of treatment with ethynodiol diacetate in “normal” women. Fertil Steril 27: 900, 1976 |
|
Spellacy WN, Buhi WC, Birk SA: Prospective studies of carbohydrate metabolism in “normal” women using norgestrel for eighteen months. Fertil Steril 35: 167, 1981 |
|
Wynn V, Godsland BA: Effects of oral contraceptives on carbohydrate metabolism. J Reprod Med 31: 892, 1986 |
|
Skouby S: Progestogens and carbohydrate metabolism: Effects at the cellular level. In Lobo RA, Whitehead MI (eds): Consensus Development Conference on Progestogens. Inter Proc J 1: 81, 1989 |
|
Neilson JH: Direct effect of gonadal and contraceptive steroids on insulin release from mouse pancreatic islets in organ culture. Acta Endocrinol 105: 245, 1984 |
|
De Pirro R, Forte F, Bertoli A et al: Changes in insulin receptors during oral contraception. J Clin Endocrinol Metab 52: 29, 1981 |
|
Luyckx AS, Gaspard UJ, Romus MA et al: Carbohydrate metabolism in women who use oral contraceptives containing levonorgestrel or desogestrel: A 6-month prospective study. Fertil Steril 45: 635, 1986 |
|
Skouby SO: Oral contraceptives: Effects on glucose and lipid metabolism in insulin-dependent diabetic women and women with previous gestational diabetes. Dan Med Bull 35: 157, 1988 |
|
Rabe T, Runnebaum B, Kohlmeyer M et al: Lipid carbohydrate and androgen metabolism in women using a triphasic oral contraceptive containing norethindrone for one year. Int J Fertil 31: 46, 1986 |
|
Knopp RH, Walden CE, Wahl PW et al: Oral contraceptive and postmenopausal estrogen effects on lipoprotein triglyceride and cholesterol in an adult female population: Relationships to estrogen and progestin potency. J Clin Endocrinol Metab 53: 1123, 1981 |
|
Spellacy WN, Ellingson AB, Tsibris JC: The effects of two triphasic oral contraceptives on carbohydrate metabolism in women during 1 year of use. Fertil Steril 51: 71, 1989 |
|
Skouby SO, Kuhl C, Molsted-Pederson L et al: Triphasic oral contraception: Metabolic effects in normal women and those with previous gestational diabetes. Am J Obstet Gynecol 153: 495, 1985 |
|
Godsland IF et al: The effects of different formulations of oral contraceptive agents on lipid and carbohydrate metabolism. N Engl J Med 323: 1375, 1990 |
|
Castelli WP: Epidemiology of coronary heart disease: The Framingham Study, Am J Med (Suppl) 2A:4, 1984 |
|
Criqui MH: Epidemiology of atherosclerosis: An updated overview. Am J Cardiol 153: 68, 1986 |
|
Miller NE, Hammett F, Saltissi S et al: Relation of angiographically defined coronary artery disease to plasma lipoprotein subfractions and apolipoproteins. Br Med J 282: 1741, 1981 |
|
Kay CR: Progestogens and arterial disease: Evidence from the Royal College of General Practitioners study. Am J Obstet Gynecol 142: 762, 1982 |
|
Schaefer EJ, Foster MD, Zech LA et al: The effects of estrogen administration on plasma lipoprotein metabolism in premenopausal females. J Clin Endocrinol Metab 57: 262, 1983 |
|
Applebaum DM, Goldberg AP, Pykalisto OJ et al: Effect of estrogen on post-heparin lypolytic activity: Selective decline in hepatic triglyceride lipase. J Clin Invest 50: 601, 1977 |
|
Kovanen PT, Brown MS, Goldstein JL: Increased binding of low density lipoprotein to liver membranes from rats treated with 17alpha-ethinyl estradiol. J Biol Chem 254: 11367, 1979 |
|
Kissebah AH, Harrigan P, Wynn V: Mechanism of hypertriglyceridemia associated with contraceptive steroids. Horm Metab Res 5: 184, 1974 |
|
Tikkanen MJ, Nikkila EA: Regulation of hepatic lipase and serum lipoproteins by sex steroids. Am Heart J 113: 562, 1987 |
|
Khokha R, Huff MW, Wolfe BM: Divergent effects of d-norgestrel on the metabolism of rat very low density and low density apoprotein B. J Lipid Res 27: 699, 1986 |
|
Krauss RM, Roy S, Mishell DM Jr et al: Effects of two low-dose oral contraceptives on serum lipids and lipoproteins. Am J Obstet Gynecol 145: 446, 1983 |
|
Percival-Smith RK, Morrison BJ, Sizto R et al: The effect of tiphasic and biphasic oral contraceptive preparations on HDL-cholesterol and LDL-cholesterol in young women. Contraception 35: 179, 1987 |
|
Capitiano GL, Bertolini S, Crosce S et al: Lipidemic changes induced by two different oral contraceptive formulations. Adv Contracept 1: 238, 1985 |
|
Gaspard UJ, Buret J, Gillian D et al: Serum lipid and lipoprotein changes induced by new oral contraceptives containing ethinylestradiol plus levonorgestrel or desogestrel. Contraception 31: 395, 1985 |
|
Ory HW, Forrest JD, Lincoln R: Making Choices-Evaluating the Health Risks and Benefits of Birth Control Methods. New York, The Alan Guttmacher Institute, 1983 |
|
Layde PM, Ory HW, Schlesselman JJ: The risk of myocardial infarction in former users of oral contraceptives. Fam Plann Perspect 14: 78, 1982 |
|
Stampfer MJ, Willet WE, Colditz GA et al: A prospective study of past use of oral contraceptive agents and risk of cardiovascular diseases. N Engl J Med 319: 1313, 1988 |
|
Further analyses of mortality in oral contraceptive users: Royal College of General Practitioners' Oral Contraceptive Study. Lancet 1:541, 1981 |
|
Adams MR, Clarkson TB, Koritnik DR et al: Contraceptive steroids and coronary artery atherosclerosis in cynomologus macaques. Fertil Steril 41: 1010, 1987 |
|
Clarkson TB, Shively CA, Morgan TM et al: Oral contraceptives and coronary atherosclerosis of cynomolgus monkeys. Obstet Gynecol 75: 217, 1990 |
|
Rossner S, Larsson-Cohn O, Carlson LA et al: Effects of an oral contraceptive agent on plasma lipids, plasma lipoproteins the intravenous fat tolerance and the post-heparin lipoprotein lipase activity. Acta Med Scand 190: 301, 1971 |
|
Fischer-Dzoga K, Wissler RW, Vesselinovitch D: The effect of estradiol on the proliferation of rabbit aortic medial tissue culture cells induced by hyperlipemic cecum. Exp Mol Pathol 39: 355, 1983 |
|
Husmann F: Long-term metabolic effects of estrogen therapy. In Greenblatt RB, Heithecker R, de Gruyter W (eds): A Modern Approach to the Perimenopausal Years: New Developments in Biosciences 2, p161. New York, DeGruyter, 1986 |
|
Milekowsky GN, Nadler J, Huey F et al: Evidence that smoking alters prostaglandin formation and platelet aggregation in women who use oral contraceptives. Am J Obstet Gynecol 159: 1547, 1988 |
|
Meade TW: Oral contraceptives clotting factors and thrombosis. Am J Obstet Gynecol 142: 758, 1982 |
|
Ambrus JL, Ambrus CM, Lillie MA et al: Effect of various estrogen treatment schedules on antithrombin III levels. Res Commun Chem Pathol Pharmacol 14: 543, 1976 |
|
Beller FK, Ebert C: Effect of oral contraceptives on blood coagulation. A review. Obstet Gynecol Surv 40: 425, 1985 |
|
Bonnar J: Coagulation effects of oral contraception. Am J Obstet Gynecol 157: 1042, 1987 |
|
Lewis MA, Heinemann LAJ et al: The increased risk of venous thromboembolism and the use of third generation progestagens. Role of Bias in Observational Research Contraception 53: 135, 1996 |
|
Food and Drug Administration, FDA Talk Paper T95-61, Rockville, MD, November 14, 1995 |
|
Vandenbroucke JP, Koster T, Briet E et al: Increased risk of venous thrombosis in oral contraceptive users who are carriers of factor V Leiden mutation. Lancet 344: 1453, 1994 |
|
Seligsohn Uri, Lubetsky A: Medical Progress: Genetic Susceptibility to Venous Thrombosis. N Engl J Med 344:1222 |
|
Vandenbroucke JP, van der Meer FJM, Helmerhorst FM et al: Factor V Leiden: Should we screen oral contraceptive users and pregnant women? BMJ 313: 1127, 1996 |
|
Vandenbroucke JP, Rosing J, Bloemenkamp KWM et al: Oral Contraceptives and the Risk of Venous Thrombosis. N Engl J Med 344: 1527, 2001 |
|
Royal College of General Practitioners: Oral Contraceptives and Health. New York, Pitman Publishing, 1974 |
|
Kaw K-T, Pearl WS: Blood pressure and contraceptive use. Br Med J 285: 403, 1982 |
|
Beck WJ Jr: Complications and contraindications of oral contraceptives. Clin Obstet Gynecol 24: 893, 1981 |
|
Wambach G: How do gestational hormones modify sodium balance? Fortschr Med 104: 561, 1986 |
|
Carr BR: Progestogens: Effect on water/salt metabolism and blood pressure. In Lobo RA, Whitehead MI (eds): Consensus Development Conference on Progestogens. Int Proc J 1: 87, 1989 |
|
Janaud A, Rouffy J, Upmalis D et al: A comparison study of lipid and androgen metabolism with triphasic oral contraceptive formulations containing norgestimate and levenorgestrel. Acta Obstet Gynecol Scand Suppl 156: 33, 1992 |
|
Ball MJ, Ahswell E, Jackson M et al: Comparison of two triphasic contraceptives with different progestogens: Effects on metabolism and coagulation parameters. Contraception 41: 363, 1989 |
|
Oelkers W, Foidart JM, Dombrovicz N et al: Effects of a new oral contraceptive containing an antimineralocorticoid progestogen, drospirenone, on the renin-aldosterone system, body weight, blood pressure, glucose tolerance, and lipid metabolism. J Clin Endocrinol Metab 80: 1816, 1995 |
|
Mishell DR, Thorneycroft IH, Nakamura RM et al: Serum estradiol in women ingesting combination oral contraceptive steroids. Am J Obstet Gynecol 114: 923, 1972 |
|
Thomas K, Ferin J: Suppression of the midcycle LH surge by a low-dose mestranol-lynestrenol oral combination. Contraception 6: 1, 1972 |
|
Mishell DR Jr, Kletzky OA, Brenner PF et al: The effect of contraceptive steroids on hypothalamic-pituitary function. Am J Obstet Gynecol 128: 60, 1977 |
|
Scott JZ, Kletzky OA, Brenner PF et al: Comparison of the effects of contraceptive steroid formulations containing two doses of estrogen on pituitary function. Fertil Steril 30: 141, 1978 |
|
Maurer RA: Estradiol regulates the transcription of the prolactin gene. J Biol Chem 157: 2133, 1982 |
|
Rigg LA, Lein A, Yen SCC: Patterns of increase in circulating prolactin levels during human gestation. Am J Obstet Gynecol 129: 454, 1977 |
|
Jacobi J, Lloyd HM, Meares JD: Induction of pituitary tumors in male rats by a single dose estrogen. Horm Metab Res 7: 228, 1975 |
|
Pituitary Adenoma Study Group: Pituitary adenomas and oral contraceptives: A multicenter case-control study. Fertil Steril 39:753, 1983 |
|
Wingrave SJ, Kay CR, Vessey MP: Oral contraceptives and pituitary adenomas. Br Med J 280: 685, 1980 |
|
Hulting AL, Werner S, Hagenfeldt K: Oral contraceptive steroids do not promote the development or growth of prolactinomas. Contraception 27: 69, 1982 |
|
Reyniak JV, Wenof M, Aubert JM et al: Incidence of hyperprolactinemia during oral contraceptive therapy, Obstet Gynecol 55:8, 1980 |
|
Cowden EA, Ratcliffe WA, Bestall GH et al: Laboratory assessment of prolactin stress. Ann Clin Biochem 16: 113, 1979 |
|
Tyson JE, Zacur H: Diagnosis and treatment of abnormal lactation. Clin Obstet Gynecol 18: 65, 1975 |
|
Goldzieher JW, Dozier TZ, Smith KD et al: Improving the diagnostic reliability of rapidly fluctuating plasma hormone levels by optimized multiple-sampling techniques. J Clin Endocrinol Metab 43: 824, 1976 |
|
Luciano AA, Sherman BM, Chapler FK et al: Hyperprolactinemia and contraception: A prospective study. Obstet Gynecol 65: 506, 1985 |
|
MacLeod SC: Endocrine effects of oral contraception. Int J Gynaecol Obstet 16: 518, 1979 |
|
Vessey MP, Lawless M, McPherson K et al: Fertility after stopping use of intrauterine contraceptive device. Br Med J 286: 106, 1983 |
|
Vessey MP, Meiser L, Flavel R et al: Outcome of pregnancy in women using different methods of contraception. Br Med J 86: 548, 1979 |
|
Jacobson C: Cytogenic study of immediate post contraceptive abortion. FDA, 1974 |
|
Harlap S, Shinono PH, Ramcharan S: Congenital abnormalities in the offspring of women who used oral and other contraceptives around the time of conception. Int J Fertil 30: 39, 1985 |
|
Metcalfe MG, Beaven DW: Plasma cortisol levels in women receiving oral contraceptive tablets. Lancet 2: 1095, 1963 |
|
Burke CW: Biologically active cortisol in plasma of oestrogen treated and normal subjects. Br Med J 2: 798, 1969 |
|
Sandberg AA, Rosenthal HE, Slaunwhite WR Jr: Certain metabolic effects of estrogen. In Salhanick HA, Kipins DM, Van de Wiele RL (eds): Metabolic Effects of Gonadal Hormones and Contraceptive Steroids. New York, Plenum Publishing, 1969 |
|
Carr BR, Parker CR Jr, Madden JD et al: Plasma levels of adrenocorticotropin and cortisol in women receiving oral contraceptive treatment. J Clin Endocrinol Metab 49: 346, 1979 |
|
Madden JD, Milewich L, Parker CR JR et al: The effect of oral contraceptive treatment on the serum concentration of dehydroepiandrosterone sulfate. Am J Obstet Gynecol 132: 380, 1978 |
|
Klove KL, Roy S, Lobo RA: The effect of different contraceptive treatments on the serum concentration of dehydroepiandrosterone sulfate. Contraception 29: 319, 1984 |
|
Thorneycroft JH, Stanczyk FZ, Bradshaw KD et al: Effect of low dose oral contraceptives on androgenic markers and acne. Contraception 60: 255, 1999 |
|
Redmond, Olson WH, Lippman JS et al: Norgestimate and ethinyl estradiol in the treatment of acne vulgaris: A randomized placebo-controlled trial. Obstet Gynecol 89: 615, 1996 |
|
Lobo RA: Hirsutism alopecia and acne. In Becker KL (ed): Principles and Practices of Endocrinology and Metabolism, p 924. Philadelphia, PA, JB Lippincott Company, 1995 |