This chapter should be cited as follows:
Fontanella F, Bilardo CM(, Glob Libr Women's Med
ISSN: 1756-2228; DOI 10.3843/GLOWM.419233
The Continuous Textbook of Women’s Medicine Series – Obstetrics Module
Volume 18
Ultrasound in obstetrics
Volume Editors:
Professor Caterina M (Katia) Bilardo, Amsterdam UMC, Amsterdam and University of Groningen, Groningen, The Netherlands
Dr Valentina Tsibizova, PREIS International School, Florence, Italy

Chapter
Ultrasound of Fetal Genitourinary Tract
First published: August 2023
Study Assessment Option
By answering four multiple-choice questions (randomly selected) after studying this chapter, readers can qualify for Continuing Professional Development points plus a Study Completion Certificate from GLOWM.
See end of chapter for details.

FETAL GENITOURINARY TRACT
Anomalies of the urinary tract constitute 20–30% of all the anomalies detected during antenatal life and have an incidence of 3 over 1000 live births.1 Urinary tract anomalies are characterized by two easily visualized sonographic markers: cystic accumulation of fluid and change in amniotic fluid. For this reason, their detection rate during fetal life is very high and reaches approximately 90%.2
Anomalies of the urinary tract include a range of anatomic and developmental abnormalities with a wide spectrum of severity and can have major consequences for the prognosis as well as for the obstetric management during pregnancy.
Normal anatomy
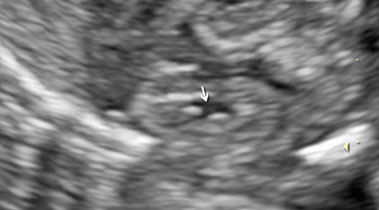
Fetal kidneys can be visualized starting from the first trimester of pregnancy. At this stage, fetal kidneys are imaged as bean-shaped structures located in the fetal renal fossae laterally to the lumbar spine. With advancing gestation, fetal kidneys become more echogenic and, starting from the third trimester of pregnancy, fetal renal calyces can be visualized (Figure 1). During the second trimester, the visualization of the kidneys can be challenging due to the fact that their echogenicity becomes similar to the one of the bowels. In this case, Doppler can be used to visualize the renal arteries.
(a) |
(b) |
1
Longitudinal section of a normal fetal kidney in the first trimester (a) and third trimester (b). In the third trimester, fetal renal calyces can be visualized.
On the upper side of each renal pole, the adrenal glands can be visualized. These appear as a rice-grain shaped organ with a hypo-echogenic ring and a central hyperechogenic line.3
Fetal urinary bladder can be visualized as soon as urinary production begins, at about 10 weeks of gestation. Fetal bladder appears as a transonic, anechoic cystic structure positioned in the fetal pelvis. Fetal urinary bladder can be distinguished from other abdominal cystic structures thanks to the two umbilical arteries, running laterally to it.
Fetal ureters and fetal urethra can only be visualized if distended and their visualization should be regarded as pathological.
Urinary tract anomalies
Malformation of the urinary tract will be discussed as follows:
- Renal anomalies
- Renal agenesis
- Hyperechogenic kidneys
- Multicystic dysplastic kidney disease
- Polycystic kidney disease
- Renal developmental variants: duplicated collecting system and ectopic kidneys.
- Obstructive anomalies
- Hydronephrosis: characteristics and differential diagnosis
- Upper urinary tract obstruction
- Lower urinary tract obstruction
- Genital anomalies: ambiguous genitalia
RENAL ANOMALIES
Renal agenesis
Renal agenesis is defined as the complete absence of kidneys and ureters. The absence of kidneys with normal ureters is called renal aplasia. This distinction cannot be done antenatally as unfilled ureters cannot be visualized echo-graphically.
Renal agenesis can be unilateral and can lead to an increased compensatory volume of the contralateral kidney and normal amount of amniotic fluid with favorable postnatal prognosis. Contrariwise, bilateral renal agenesis entails an invariably lethal outcome with severe oligo- and anhydramnios starting from the second trimester and subsequent pulmonary hypoplasia and contractures with the typical Potter sequence (retrognathia, club foot, and limb contractures).
Epidemiology
Unilateral renal agenesis has an incidence of 1/1000.3 The recurrence risk is 4–9%.4,5,6 Bilateral agenesis can be isolated or associated with either isolated structural anomalies or in the context of a syndrome, for instance, as part of the VACTERL spectrum.7
Ultrasound
The diagnosis of bilateral renal agenesis should be considered in the case of absent urinary bladder filling, oligo- or anhydramnios and impossibility in visualizing fetal kidneys. The ultrasound imaging is often suboptimal due to severe oligohydramnios. In this case, color or power Doppler can help in confirming the absence of both renal arteries and in identifying the superior vesical arteries surrounding the bladder with absence of bladder filling. Premature rupture of membranes and severe growth restriction should be considered in the differential diagnosis, although in both these cases, contrarily to bilateral renal agenesis, bladder filling is present. On the other hand, a scarce bladder filling cannot totally exclude renal agenesis as a rudimentary cystic bladder can be present.8
Another possible pitfall in prenatal diagnosis is confounding the adrenal glands with kidneys. However, these two organs present a different shape: adrenal glands appear normally triangular, while kidneys present as bean-shaped structure. In the case of renal agenesis, adrenal glands tend to be prolapsed and occupy the renal fossa, appearing with a rounder and more elongated shape, named the ‘’lying down’’ adrenal sign.9
Prognosis and management
Bilateral renal agenesis is a lethal condition. The prognosis of unilateral renal agenesis is generally favorable with compensatory hypertrophy of the contralateral kidneys and normal renal function postnatally.10
Hyperechogenic kidneys
Multicystic dysplastic kidney disease
Multicystic dysplastic kidney (MCKD) is a form of renal dysplasia and cystic degeneration associated with an atretic ureter.
Epidemiology
MCKD is in most of the cases (80%) unilateral and can be associated with other structural anomalies.11 If not isolated, there is an increased risk of chromosomal anomalies, mainly trisomy 18, trisomy 13, and Meckel Gruber syndrome. Meckel Gruber syndrome is characterized by bilateral multicystic kidneys with small uniform cysts, postaxial polydactyly, and encephalocele. Fetuses with Meckel Gruber syndrome present a normal karyotype.12
Ultrasound diagnosis
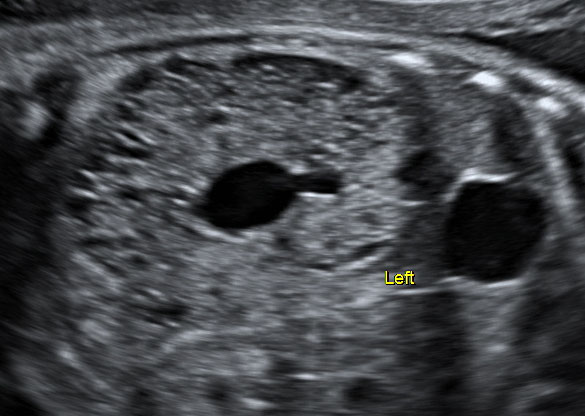
Multicystic kidneys are characterized by hyperechogenic parenchyma filled with multiple non-communicating cysts of variable sized (Figure 2) and kidneys often presenting an increased volume (Figure 2). In 30% of the cases, MCKD can lead to oligohydramnios, absence of bladder filling and renal insufficiency, while a normal amount of amniotic fluid suggests normal renal function in the contralateral kidney. Cysts can increase in size and subsequently regress during pregnancy. The prognosis of a unilateral MCKD without associated anomaly is favorable.11
2
Fetus with multicystic dysplastic kidney at 26 weeks of gestation.
Polycystic kidney disease
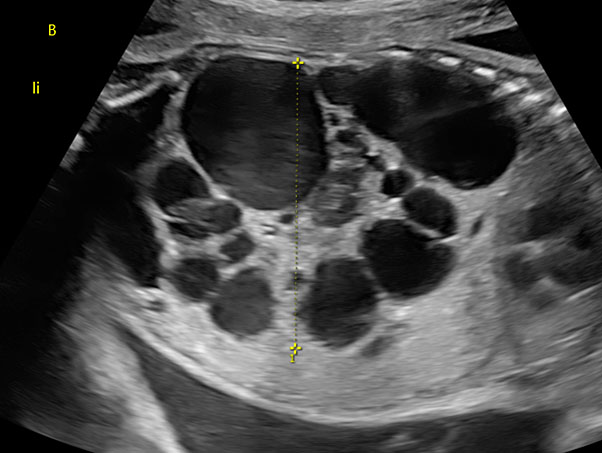
Polycystic kidney disease occurs in two forms of genetical disorders characterized by inherited cystic renal anomalies (Figure 3).13
3
Fetus with polycystic dysplastic kidney at 28 weeks of gestation.
Two forms of polycystic kidney dysplasia can be distinguished:
- Autosomal recessive: ARPKD. ARPKD is caused by a mutation of PKHD1 gene on chromosome 6p21.14 ARPKD is characterized by bilateral enlarged and hyperechogenic kidneys, absent bladder filling, and usually severe oligohydramnios. More than 90% of the renal tissue is cystic and not functional, leading to oligo- or anhydramnios early during pregnancy and to long hypoplasia. ARPKD has an incidence of 1/20.000–40.000. Prognosis is poor with up to 40–50% of the affected individuals dying in the early neonatal period, while live-born infants have a high-risk of progressing to end-stage renal disease.13,15
- Autosomal dominant: ADPKD. ADPKD is caused by a mutation in PKD1 gene on chromosome 16 or PKD2 on chromosome 4 and represents the most common hereditary kidney disorder with an incidence of 1/1000.14 The condition is inherited with an autosomal dominant pattern. Typical is the adult onset, with symptoms usually presenting in the third to fifth decades of life. In fetal life, ADPKD can show an abnormal aspect of fetal kidneys in the third trimester of pregnancy. Kidneys appear enlarged and with increased parenchymal echogenicity mainly in the outer layer due to the presence of microcysts. Bladder filling and amount of amniotic fluid is usually normal.16 Subjects with ADPKD have generally normal life expectancy, although evidence of disease yet during fetal life could entail a poorer prognosis. The cysts can also be present in the liver leading to polycystic liver and the need for surgical intervention.13 In the case of suspected ADPKD in the fetus, it is advised to offer further examinations to the parents.8
Renal developmental variants: duplicated collecting system and ectopic kidney
Duplicated collecting system is characterized by the presence of two ureters. This can be partial if the ureters are fused together before connecting to the bladder, or complete if both ureters will separately merge into the bladder. The double system occurs in 0.7–4% of the population and more frequently in women than men (Video 1).17
1
Duplicated collecting system.
At ultrasound examination, two separated dilated renal pelvis can be visualized in one kidney, which can clearly appear bigger than the contralateral kidney (Figure 4). The upper pole of the kidney often presents an abnormal inserted ureter with subsequent dilatation of the collecting system and hydronephrosis. The prognosis is generally good, unless a severe obstruction occurs resulting in dysplastic changes. In the case of recurrent urinary infection postnatally, surgery can be indicated.18
|
|
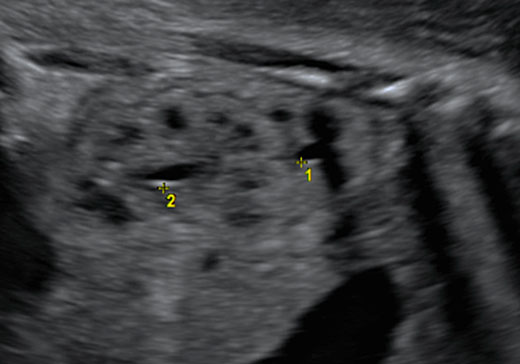
4
Fetus with duplicated collecting system at 32 weeks of gestation.
An ectopic kidney can be suspected in the case of empty renal fossa with normal aspect of contralateral kidney without compensatory changes.19 The ectopic kidney can be visualized as a pelvic mass, which need to be differentiated from an ovarian cyst, obstructed bowel or sacrococcygeal teratoma. In some cases, the actual kidney cannot be timely identified until the third trimester. Ectopic kidneys can be isolated or associated with other congenital anomalies or can be even associated with vesico-ureteral reflux or multicystic dysplastic kidney. The prognosis is favorable, particularly if the anomaly is isolated.
OBSTRUCTIVE ANOMALIES OF THE URINARY TRACT
Hydronephrosis: characteristics and differential diagnosis
Fetal pelvis can be evaluated in a sagittal section with the spine at 6 or 12 o’clock and by measuring the pelvis diameter placing the callipers perpendicularly between renal pelvis and renal parenchyma junction. Fetal antero-posterior pelvic diameter is considered normal if it is smaller the 4 mm between 16 and 27 weeks of gestation and smaller than 7 mm at and after the 28th week of gestation.20,21 Recently there is agreement in defining, as dilated, a renal pelvis of equal or more than 7 mm in diameter at the mid-trimester examination.22,23
Fetal hydronephrosis occurs in 1–2% of all pregnancies and is more common in male fetuses, with a male–female ratio of 2 : 1.24,25 In 50–70% of the cases, fetal hydronephrosis is a transient ultrasound sign without relevant clinical implications. However, it can also be associated with vesico-ureteral reflux, obstructive anomalies of the urinary tract or considered as a minor marker of trisomy 21. The most common cause of persistent and not physiologic fetal hydronephrosis is an ureteropelvic junction obstruction (10–30% of the cases) followed by vesico-ureteral reflux (10–40%).24,25,26
A follow-up ultrasound scan is advised at around 32 weeks of gestation as most of the cases will present a spontaneous resolution before this gestational age. In the case of progression, an obstructive anomaly of the urinary tract should be considered.26
Obstructive anomalies
The obstructive anomalies of the urinary tract can be categorized in two groups according to the localization of the obstruction:
- Upper urinary tract obstruction (UUTO)
- Lower urinary tract obstruction (LUTO)
Epidemiology
Congenital urinary tract obstructions represent the biggest group of urological anomalies and the most common cause of renal transplant during childhood.27,28
In UUTO, the obstruction occurs at the level of renal pelvis or ureters, often leading to unilateral hydronephrosis, while in LUTO the obstruction occurs below the bladder neck leading typically to bilateral hydronephrosis.
Upper urinary tract obstructions
Etiology
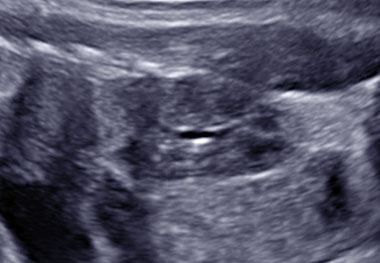
Uretero-pelvic junction (UPJ) obstruction is the most common lesion among this group, with an incidence of 1/5000–20,000, followed by uretero-vesical obstruction, which can be caused by ureteric strictures, ureteric atresia, primary megaureter, vascular obstruction, diverticulum, valves, retrocaval ureter, ureterocele, and vesicoutereral reflux.29 These causes of upper urinary tract obstruction can be either unilateral or bilateral (Video 2).
2
Megaureter left.
Ultrasound appearance and natural history
Hydronephrosis is a typical sign of upper urinary tract obstruction, although most of the hydronephrosis can be transient and functional and without evidence of urinary obstruction. In unilateral UPJ obstructions, the hydronephrosis is typically unilateral with dilated infundibulum. Dilatation of the calices can also be seen. The bladder is not dilated and does not present a thickened wall. The most severe forms of UPJ obstruction can lead to massive dilatation of the upper urinary tract with rupture of the collecting system and perinephric urinoma or urinary ascites.2
Prognosis
The prognosis of upper urinary tract obstruction is generally favorable unless other severe associated congenital anomalies are diagnosed.
Lower urinary tract obstructions
Etiology
In the case of LUTO, the obstruction occurs below the level of the bladder neck. LUTO is in most of the cases caused by the occurrence of posterior urethral valves blocking partially or totally fetal urination.29,30 This typically occurs in male fetuses. LUTO can also be caused by urethral atresia (mainly in female fetuses with other associated congenital anomalies) and urethral strictures.31
Ultrasound
The characteristic antenatal presentation of congenital LUTO include a dilated urinary bladder (also called megacystis), bilateral hydronephrosis and, after the second trimester of pregnancy, oligohydramnios.29,32
LUTO can also be associated with other congenital anomalies, mainly hearth and spinal defects, as well as with chromosomal anomalies. For this reason, when LUTO is suspected, a detailed antenatal morphological ultrasound scan should be performed and fetal karyotyping should be offered.33 Congenital syndromes should also be included in the differential diagnosis, such as megacystis-microcolon-intestinal hypoperistalsis syndrome, a very rare syndrome characterized by a largely dilated non-obstructed megacystis with very small colon and decreased of absent intestinal peristalsis.33
Fetal sex, presence of associated anomalies, gestational age at occurrence of megacystis and oligohydramnios can strongly help in the differential diagnosis and counselling. In particular, a series of antenatal characteristics can suggest a poor prognosis, such as female sex, megacystis occurring during the first trimester of pregnancy, the presence of other associated anomalies and the occurrence of early oligohydramnios, in particular, before the 26th week of gestation.32,33,34
Prognosis
If untreated, fetal LUTO entails a mortality rate of 45% and among the live-born neonates around 25–30% will develop an end-stage chronic renal impairment requiring dialysis or transplantation during childhood.35,36 The prognosis of LUTO strongly depends on its underlying etiology.37,38 In fact, fetuses with urethral atresia present a very poor prognosis with severe megacystis and early anhydramnios, while obstructions caused by urethral valves can also be partial and manifest only later during fetal life, thus entailing a better prognosis.39 To date, an accurate prediction of postnatal prognosis using antenatal ultrasound is still challenging.
AMBIGUOUS GENITALIA
The term ‘ambiguous genitalia’ encompasses a group of anomalies where the external genitalia are not surely suggestive of a male or female phenotype, or whether the external genitalia are different from the genetic sex.40 The incidence of ambiguous genitalia is around 1–5000 livebirths.41
Etiology
The most frequent anomaly detected in male fetuses are micropenis, ventral curvature of the penis, retained testes, scrotum bifidum, and hypospadias. While female fetuses can present clitoral hypertrophy.
Ambiguous genitalia can also be caused by endocrine disorders, such as congenital adrenal hyperplasia (CAH). CAH is caused by deficiency of 21-hydroxylase and constitutes the most common cause of virilization in female fetuses.2 Moreover, a variety of congenital syndromes can be characterized by ambiguous genitalia, including Robinow syndrome (short stature, short arms, and characteristic facies), Opitz syndrome (hypertelorism and hypospadias in male fetuses), Aarskog syndrome (X-linked disorder with short stature, hypertelorism, short nostrils, and genitalia dysplasia), Smith Lemli Opitz syndrome (intrauterine growth restriction, holoprosencephaly and cleft palate), WAGR (Wilms tumor, aniridia, growth restriction, and genital anomalies) or Prader Willi syndrome. The risk of associated chromosomal anomalies is relatively low. The most common cause of hypoplastic male genitalia is severe fetal growth restriction. If the post-natal outcome is favorable, the condition usually spontaneously improves during life.42
Ultrasound
Antenatal differential diagnosis of ambiguous genitalia is extremely challenging with high rate of false-positive results, as for example, a micropenis with cryptorchidism cannot always be clearly differentiated from clitoral hypertrophy.42 Anomalies of the sex organs are sometimes combined with malformations of the urinary tract.43 Facial clefts and congenital heart disease can also be founded.
Management and prognosis
Prognosis and postnatal quality of life strongly depends on the underlying etiology. In the case of CAH, treatment of the fetus and neonate should be promptly discussed in a multidisciplinary team together with pediatricians, endocrinologists, and genetics.
PRACTICE RECOMMENDATIONS
Fetal urinary bladder and fetal kidneys can be visualized starting from the first trimester of pregnancy.
- Renal agenesis is a lethal condition if bilateral, while a unilateral renal agenesis entails generally a favorable prognosis.
- Multicystic kidneys are characterized by multiple non-communicating cysts and an increased volume of the kidneys. Cysts can increase in size and subsequently regress during pregnancy.
- Polycystic kidney diseases are two forms of genetical disorders characterized by cystic renal anomalies with an inherited pattern and present a different prognosis.
- Fetal hydronephrosis occurs in 1–2% of all pregnancies and in 50–70% of the cases will regress spontaneously before 32 weeks of gestation. A follow-up scan at 32 weeks of gestation is therefore recommended.
- The most common cause of obstructive uropathy and non-physiological hydronephrosis is ureteropelvic junction obstruction.
- A lower urinary tract obstruction (LUTO) should be suspected in the case of dilated urinary bladder (megacystis) associated with bilateral hydronephrosis.
- LUTO can also be associated with other congenital anomalies as well as with chromosomal anomalies. Therefore, a detailed antenatal morphological ultrasound is advised and fetal karyotyping should be offered.
- Fetal LUTO entails a high risk of neonatal death (45%) and poor postnatal outcome with end-stage chronic renal impairment.
- Male fetuses with LUTO suspected later in pregnancy and a normal amount of amniotic fluid until 26 weeks entails a more favorable prognosis.
CONFLICTS OF INTEREST
The author(s) of this chapter declare that they have no interests that conflict with the contents of the chapter.
Feedback
Publishers’ note: We are constantly trying to update and enhance chapters in this Series. So if you have any constructive comments about this chapter please provide them to us by selecting the "Your Feedback" link in the left-hand column.
REFERENCES
Tain YL, Luh H, Lin CY, et al. Incidence and Risks of Congenital Anomalies of Kidney and Urinary Tract in Newborns A Population-Based Case-Control Study in Taiwan. [cited 2022 Dec 28]; Available from: www.md-journal.com. | |
Diana W. Bianchi TMCMEDAFM. Fetology, 2nd edn. Vol. 9780071442015. Medicine & Health Science Books. | |
Jeanty P, Chervenak F, Grannum P, et al. Normal ultrasonic size and characteristics of the fetal adrenal glands. Prenat Diagn 1984;4(1):21–8. | |
Carter C0, Evans K, Pescia’ G. A family study of renal agenesis. J Med Genet 1979;16:176–88. | |
McPherson E, Carey J, Kramer A, et al. Dominantly inherited renal adysplasia. Am J Med Genet [Internet] 1987;26(4):863–72. | |
Roodhooft AM, Birnholz JC, Holmes LB. Familial nature of congenital absence and severe dysgenesis of both kidneys. N Engl J Med [Internet] 1984;310(21):1341–5. | |
Sanna-Cherchi S, Weng PL, Scolari F, et al. Genetic approaches to human renal agenesis/hypoplasia and dysplasia. Available from: http://www.marchofdimes.com. | |
Adama van Sheltema P, Oepkes D. Aangeboren afwijkingen van de nieren en urinewegen. Echoscopie in de Verloskunde en Gynaecologie, 5th edn. 2016. | |
Hoffman CK, Filly RA, Callen PW. The “lying down” adrenal sign: a sonographic indicator of renal agenesis or ectopia in fetuses and neonates. Journal of Ultrasound in Medicine 1992;11(10):533–6. | |
Talati AN, Webster CM, Vora NL. Prenatal genetic considerations of congenital anomalies of the kidney and urinary tract (CAKUT). | |
Hains DS, Bates CM, Ingraham S, et al. Management and etiology of the unilateral multicystic dysplastic kidney: A review. Pediatric Nephrology 2009;24(2):233–41. | |
Hartill V, Szymanska K, Sharif SM, et al. Meckel-Gruber Syndrome: An Update on Diagnosis, Clinical Management and Research Advances. Front Pediatr 2017;5. | |
Bergmann C, Guay-Woodford LM, Harris PC, et al. Polycystic kidney disease. Nat Rev Dis Primers 2018;4(1):50. | |
Cornec-Le Gall E, Torres VE, Harris PC. BRIEF REVIEW Genetic Complexity of Autosomal Dominant Polycystic Kidney and Liver Diseases. J Am Soc Nephrol 2018;29:13–23. | |
Zerres K, Mücher G, Becker J, et al. Prenatal diagnosis of autosomal recessive polycystic kidney disease (ARPKD): molecular genetics, clinical experience, and fetal morphology. Am J Med Genet 1998;76(2):137–44. | |
MacDermot KD, Saggar-Malik AK, Economides DL, et al. Prenatal diagnosis of autosomal dominant polycystic kidney disease (PKD1) presenting in utero and prognosis for very early onset disease. J Med Genet 1998;35(1):13–6. | |
Hopkins LM, for Maternal-Fetal Medicine S. Duplicated collecting system. The American Journal of Obstetrics & Gynecology 2021;225:B12–3. | |
Didier RA, Chow JS, Kwatra NS, et al. The duplicated collecting system of the urinary tract: embryology, imaging appearances and clinical considerations. Pediatr Radiol 2017;47(11):1526–38. | |
Meizner I, Yitzhak M, Levi A, et al. Fetal pelvic kidney: a challenge in prenatal diagnosis? Ultrasound Obstet Gynecol 1995;5(6):391–3. | |
Chitty LS, Altman DG. Charts of fetal size: kidney and renal pelvis measurements. Prenat Diagn 2003;23(11):891–7. | |
Scott JES, Renwick M. Antenatal renal pelvic measurements: what do they mean? BJU Int 2001;87(4):376–80. | |
Nguyen HT, Herndon CDA, Cooper C, et al. The Society for Fetal Urology consensus statement on the evaluation and management of antenatal hydronephrosis. J Pediatr Urol 2010;6(3):212–31. | |
Ismaili K, Hall M, Donner C, et al. Results of systematic screening for minor degrees of fetal renal pelvis dilatation in an unselected population. Am J Obstet Gynecol 2003;188(1):242–6. | |
Liu DB, Armstrong WR, Maizels M. Hydronephrosis: Prenatal and Postnatal Evaluation and Management. Clin Perinatol 2014;41(3):661–78. | |
Elder JS. Antenatal Hydronephrosis: Fetal and Neonatal Management. Pediatr Clin North Am 1997;44(5):1299–321. | |
Feldman DM, DeCambre M, Kong E, et al. Evaluation and follow-up of fetal hydronephrosis. J Ultrasound Med 2001;20(10):1065–9. | |
Denny E, Quinlan-Jones E, Bibila S, et al. The experience of pregnant women with a diagnosis of fetal lower urinary tract obstruction (LUTO). Midwifery 2014;30(6):636–42. | |
Parkhouse HF, Woodhouse CR. Long-term status of patients with posterior urethral valves. Urol Clin North Am 1990;17(2):373–8. | |
Cheung KW, Morris RK, Kilby MD. Congenital urinary tract obstruction. Best Pract Res Clin Obstet Gynaecol 2019;58:78–92. | |
SJ H, B P, G M, A A. Posterior urethral valves. ScientificWorldJournal 2009;9:1119–26. | |
Gonzalez R, Filippo R de, Jednak R, et al. Urethral atresia: long-term outcome in 6 children who survived the neonatal period. J Urol 2001;165:2241–4. | |
Fontanella F, Duin LK, Adama van Scheltema PN, et al. Prenatal diagnosis of LUTO: how to improve diagnostic accuracy. Ultrasound in Obstetrics & Gynecology 2018;1–20. | |
Fontanella F, Maggio L, Verheij JBGM, et al. Fetal megacystis: a lot more than LUTO. Ultrasound Obstet Gynecol 2018. | |
Kohl T. Management of very young fetuses with LUTO. Nat Rev Urol 2022;19(10):627–8. | |
Morris RK, Kilby MD. An overview of the literature on congenital lower urinary tract obstruction and introduction to the PLUTO trial: Percutaneous shunting in lower urinary tract obstruction. Australian and New Zealand Journal of Obstetrics and Gynaecology 2009;49(1):6–10. | |
Morris RK, Kilby MD. Congenital urinary tract obstruction. Best Pract Res Clin Obstet Gynaecol 2008;22(1):97–122. | |
Haeri S. Fetal Lower Urinary Tract Obstruction (LUTO): a Practical Review for Providers, 2015. | |
Saccone G, D’Alessandro P, Escolino M, et al. Antenatal intervention for congenital fetal lower urinary tract obstruction (LUTO): a systematic review and meta-analysis. J Matern Fetal Neonatal Med 2020;33(15):2664–70. | |
Fontanella F, van Scheltema PNA, Duin L, et al. Antenatal staging of congenital lower urinary tract obstruction. Ultrasound Obstet Gynecol [Internet] 2019;53(4):520–4. | |
Makiyan Z. Systematization of ambiguous genitalia. Organogenesis 2016;12(4):169. | |
Kutteh WH, Santos‐Ramos R, Ermel LD. Accuracy of ultrasonic detection of the uterus in normal newborn infants: implications for infants with ambiguous genitalia. Ultrasound Obstet Gynecol 1995;5(2):109–13. | |
Dario Paladini, Paolo Volpe. Ultrasound of Congenital Fetal Anomalies: Differential Diagnosis and Prognostic Indicators, 2nd edn. 2014. | |
Aarskog D. Syndromes and Genital Dysmorphology. Horm Res Paediatr 1992;38(Suppl. 2):82–5. |
Online Study Assessment Option
All readers who are qualified doctors or allied medical professionals can automatically receive 2 Continuing Professional Development points plus a Study Completion Certificate from GLOWM for successfully answering four multiple-choice questions (randomly selected) based on the study of this chapter. Medical students can receive the Study Completion Certificate only.
(To find out more about the Continuing Professional Development awards programme CLICK HERE)